
Remember me

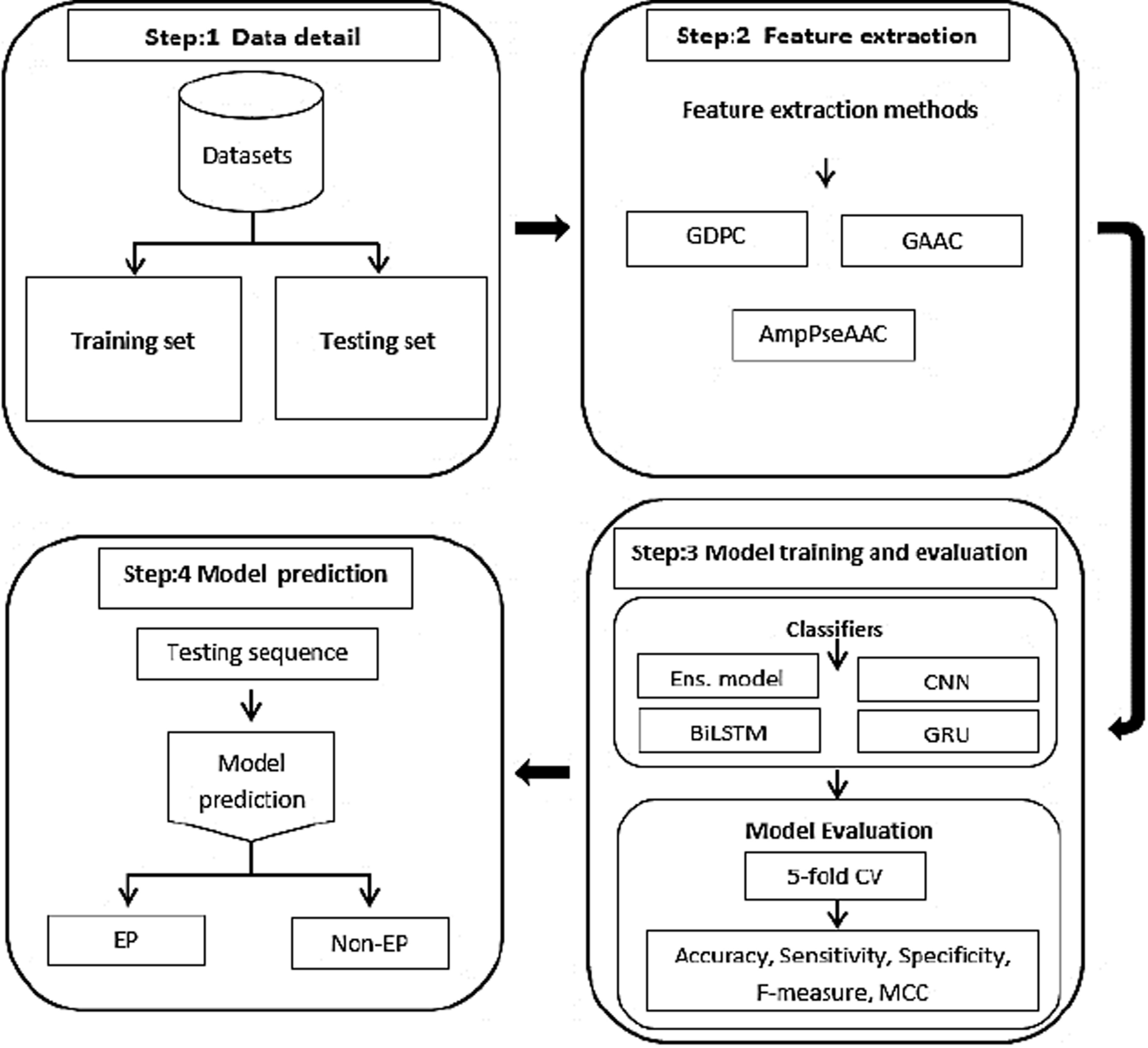
To illustrate the methodology adopted in this study, the proposed framework is introduced in Fig. 1. In addition the general framework is illustrated in detail to provide a thorough grasp of the process. Each of the pipeline’s six consecutive steps ensures reliable feature selection and categorization while also contributing to the overall analysis process. A detailed explanation of each step is presented below:
Data Collection and Preprocessing in the first step, a newly compiled genetic dataset focusing on Non-Hodgkin Lymphomas (NHL) and related rheumatologic disorders is gathered from public sources like NCBI. Preprocessing includes cleaning the data and selecting relevant features for further analysis.
Feature Encoding (Tripeptide Composition) in this stage, the Tripeptide Composition (TPC) approach is used to convert protein sequences into numerical representations. To make sure that each protein sequence is compatible with machine learning methods, it is transformed into an 8000-dimensional feature vector.
Mutual Information Computation and Feature Clustering every feature pair’s Mutual Information (MI) is computed in order to evaluate its correlation. Features are then grouped according to their similarity using k-means clustering. Only non-redundant features are chosen for additional analysis thanks to this clustering process, which also helps to reduce redundancy.
Feature Selection using Clustered-Based Binary Grey Wolf Optimizer (CB-BGWO) each feature cluster is subjected to the CB-BGWO algorithm, which determines which subset of characteristics is the most informative. By reducing duplication and increasing relevance, this novel optimization method iteratively improves the feature selection procedure.
Classification using Multiple Classifiers Gradient Boosting Classifier, Gaussian Process Classifier, Support Vector Classification (SVC), Logistic Regression, and Stochastic Gradient Descent (SGD) Classifier are among the classifiers that employ the reduced feature set as input. The prediction performance of each classifier is evaluated through training and testing.
Performance Evaluation and Analysis several metrics, including accuracy, precision, recall, F1-score, and Area Under the ROC Curve (AUC), are used to assess each classifier’s performance. The suggested framework’s robustness and dependability are guaranteed by this thorough assessment.
Fig. 1
Proposed framework pipeline for feature selection and classification using CB-BGWO
DatasetBuilding a robust dataset is a key component of our research, which attempts to elucidate the genetic connection between Non-Hodgkin Lymphomas (NHL) and associated rheumatologic disorders. To guarantee methodical selection and reduce subjective interference, the dataset-gathering and refining process was carried out using a carefully constructed pipeline. This pipeline was divided into several phases, including the initial identification of the dataset, the definition of the selection criteria, and the use of computational techniques to expand the gene set.
Using public resources such as PubMed, Gene, and NCBI Protein, a thorough literature study was conducted to assemble the first dataset. To guarantee high reliability and relevance, genes linked to Sjögren’s syndrome, NHL, systemic lupus erythematosus (SLE), and celiac disease were found in peer-reviewed publications. We only considered genes that have been implicated in numerous research and found to have a major influence on these illnesses. An exhaustive list of 189 genes has been identified in recent scientific studies. The diseases NHL (54 genes), SLE (25 genes), CD (77 genes), and Sjögren’s syndrome (33 genes) are linked to these genes. For instance, because of their frequent associations in scientific literature, genes like HLA-DRB1, TNFSF4, and IRF5 were picked for SLE, while genes like LCE2B, KNG1, and TG were chosen for NHL. Table 1 provides a detailed summary of these genes, their associated diseases, and references from the literature.
GeneMANIA [105] was used to find more first- and second-degree interacting genes in order to increase the dataset’s comprehensiveness specifically specified. We were able to expand the dataset using this computational method by adding biologically significant genes that are closely linked by molecular pathways but may not have been specifically identified in individual research. Consequently, we produced a more comprehensive dataset that encompasses a wider genetic landscape. As a result, the number of genes in the dataset increased from the initially identified set to a total of 229 genes. The detailed procedure for creating the extensive genetic dataset is shown in the flowchart in Fig. 2
Fig. 2
Proposed framework pipeline for dataset generation
A wide representation of the genetic landscape is guaranteed by this carefully curated and varied dataset, which contains genes from several autoimmune and rheumatologic disorders in addition to increased gene interactions via computational techniques. The dataset’s extensive coverage of biologically critical genes and molecular pathways makes it applicable to a wider range of study contexts in addition to being extremely reliable for comprehending the genetic basis of Non-Hodgkin Lymphomas (NHL) and related illnesses. This wider applicability makes it easier to create more generalized models for genetic study and increases the possibility of cross-disease insights. In addition its strong capability our framework viable candidate for real world usage.
Table 1 Genes associated between non-Hodgkin lymphomas and rheumatologic diseasesData preprocessingTo guarantee the accuracy, consistency, and dependability of the protein sequence dataset, a number of crucial data pretreatment procedures were carried out before the Tripeptide Composition (TPC) approach was applied. These actions were necessary to standardize the dataset for further analysis, remove redundancy, and reduce noise.
To prevent mistakes during feature extraction, the dataset was first inspected for missing or partial protein sequences, and such entries were eliminated. To guarantee that every protein sequence in the dataset was distinct and did not add bias into the study, duplicate sequences were found and removed.
A thorough analysis conducted in the study’s first phase found 238 genes linked to Non-Hodgkin Lymphomas (NHL) and other rheumatologic conditions. Because their amino acid sequences were less than 20 amino acids, nine of these genes were disqualified, potentially jeopardizing the feature extraction process’s dependability. As a result, 229 genes were included in the modified dataset.
After each gene was chosen, numerical characteristics were extracted from its amino acid sequences using the Tripeptide Composition (TPC) approach. In order to ensure interoperability with machine learning models, our procedure transformed each protein sequence into an 8000-dimensional feature vector. Important metrics including mean, median, standard deviation, and variance were computed in order to assess the statistical characteristics of the features that were extracted. According to the findings, the dataset is roughly normally distributed, with a mean value of 0.527 ± 0.02 and a median of 0.525 for the features. The recovered features are well-clustered around the mean, which lowers noise and increases the machine learning models’ dependability, as indicated by the standard deviation of 0.081. Additionally, the variance was 0.0066, indicating that the dataset is resilient and appropriate for additional research because it does not contain extreme outliers.
To make classification easier, each protein sequence was then labeled with the disease class that corresponded to it.
These preprocessing procedures were essential for improving the analysis’s resilience and guaranteeing that the TPC approach worked with a clear, organized dataset, which produced more accurate and dependable results.
Feature encodingDue to the large range of amino acids and the distinct structures and functions of proteins, protein feature extraction is a more challenging task than DNA and RNA sequencing. Various techniques for feature extraction have been put forth throughout time to address this complexity [110].
We have prepared the protein sequence data for computer analysis by encoding it using the Composition of Tripeptide Composition (TPC). This enhanced approach increases our analysis and offers a comprehensive perspective of the properties associated with proteins that are vital to comprehending these disorders.
Three consecutive native amino acids combine to generate TPC, an efficient and minimum biological recognition signal. This signal provides an invaluable model for the identification of peptides and other small biological compounds that behave as biological function modulators [100]. Tripeptides have been shown to be useful in de novo protein design and in the prediction of palatable oligopeptide structures by Anishetty et al. [8]. The TPC yielded 8000-dimensional feature vectors. A vector is defined as a sequence of proteins:
$$} = [\alpha _, \alpha _, \ldots , \alpha _, \ldots , \alpha _]^,$$
(1)
where \(T\) denotes the transpose of the vector and \(\alpha _\) represents the frequency of the \(i\)th tripeptide for \(i = 1,2,3, \ldots , 8000\). The frequency \(\alpha _\) can be expressed as:
$$\alpha _ = \frac}.$$
(2)
Here, \(D_\) and \(L\) denote the frequency of the \(i\)-th tripeptide and the length of the protein chain, respectively.
With the use of this quantitative measure, we can convert protein sequences into a numerical format that is appropriate for machine learning algorithms, making it easier to anticipate and analyze the properties and activities of proteins.
Feature similarity analysis using mutual informationIn this study, we utilize the Mutual Information (MI) principles to determine the level of correlation between various features in our dataset. MI is a reliable metric for calculating how dependent variables are on one another. In essence, it expresses how knowing about one trait reduces uncertainty about another. This work uses MI to determine each feature’s importance with respect to the target class and to evaluate how similar two feature pairs are to one another.
The quantity of information learned about one feature by watching the other is quantified by the Mutual Information (MI) between two feature vectors, \(f_\) and \(f_\). It is defined as the product of the marginal distributions \(p(f_)\) and \(p(f_)\), and the relative entropy between the joint distribution \(p(f_, f_)\). It is represented as follows:
$$I(f_; f_) = \sum _ \in f_} \sum _ \in f_} p(f_, f_) \log \left( \frac, f_)})p(f_)}\right) ,$$
(3)
where:
\(p(f_, f_)\) is the joint probability distribution function of \(f_\) and \(f_\),
\(p(f_)\) and \(p(f_)\) are the marginal probability distribution functions of \(f_\) and \(f_\) respectively,
\(f_\) and \(f_\) are vectors representing the sets of all possible values for the respective features,
The logarithm is typically taken to base 2 if the information is measured in bits, or to the base e (natural logarithm) for nats.
MI is a non-negative, symmetric function such that \(I(f_; f_) = I(f_; f_)\), and it only has a zero value if and only if \(f_\) and \(f_\) are independent. The degree to which uncertainty is reduced increases with the value of MI. MI can alternatively be stated as follows in terms of entropy:
$$I(f_; f_) = H(f_) + H(f_) - H(f_, f_),$$
(4)
where:
\(H(f_)\) and \(H(f_)\) are the marginal entropies of \(f_\) and \(f_\),
\(H(f_, f_)\) is the joint entropy of \(f_\) and \(f_\).
The mutual information can also be interpreted as the expected value of the pointwise mutual information (PMI):
$$\begin I(f_; f_)= & }_,f_)}[\text (f_; f_)] \\= & }_,f_)}\left[ \log \left( \frac, f_)})p(f_)}\right) \right] , \end$$
(5)
where \(}\) denotes the expectation operator.
Binary grey wolf optimization (bGWO)The Binary Grey Wolf Optimization (bGWO) extends the continuous grey wolf optimization (CGWO) to address discrete problem spaces, such as those encountered in feature selection tasks. Solution vectors in bGWO are binary and limited to the points on a hypercube that symbolize the search space. Attraction to the alpha (\(\alpha\)), beta (\(\beta\)), and delta (\(\delta\)) wolves update each wolf’s location, signifying the top three solutions found so far [29].
The update equation for each wolf is guided by the position vectors \(x_\), \(x_\), and \(x_\) and is given by:
$$X^_ = }(x_, x_, x_),$$
(6)
where a crossover operation between binary solutions x, y, z is indicated by the notation Crossover(x, y, z). The influence of the alpha, beta, and delta wolves on the ith wolf’s movement is represented by the vectors \(x_, x_, x_\), which are defined as follows:
$$x_^ = 1 & } (x_^ + }_^) \ge 1, \\ 0 & }, \end\right. }$$
(7)
where \(x_^\) is the alpha wolf’s position vector in dimension d, and \(}_^\) is a binary step calculated as:
$$}_^ = 1 & } }_^ \ge }, \\ 0 & }, \end\right. }$$
(8)
where \(}_^\) is a continuous-valued step size in dimension d defined by a sigmoidal function, and rand is a random number drawn from a uniform distribution:
$$}_^ = \frac^D_^ - 0.5)}}.$$
(9)
\(D_^\) and \(A_^\) have values that are obtained from other equations in the system.
To achieve bGWO, the alpha, beta, and delta solution contributions are combined using a stochastic crossover technique. For every dimension, the crossover output is ascertained by:
$$x_ = a_ & } \le \frac, \\ b_ & } \frac < } \le \frac, \\ c_ & }, \end\right. }$$
(10)
where the binary values for the first, second, and third solutions in dimension d are \(a_\), \(b_\), and \(c_\), respectively, and the rand is once more a random number from a uniform distribution.
Finding a subset of features that offers the optimal trade-off between classification performance and the number of selected features is the aim of feature selection in classification tasks, where the bGWO strategy proves to be very beneficial.
k-Means clusteringA common technique for clustering in a number of disciplines, such as bioinformatics, computer vision, and data mining, is the k-means algorithm. To minimize the within-cluster sum of squares (WCSS), k-Means aims to divide a set of N data points into K unique non-overlapping subgroups, or clusters.
k-Means clustering attempts to divide the N observations into \(K \,(\le N)\) sets \(S = \, S_, \ldots , S_\}\) in order to minimize the within-cluster variance given a set of observations \((x_, x_, \ldots , x_)\), where each observation is a d-dimensional real vector:
$$} \min _ \sum _^ \sum _} ||x - \mu _||^,$$
(11)
where \(\mu _\) is the mean of points in \(S_\).
The algorithm can be summarized in the following steps:
1.Initialization: Randomly select K data points as the initial centroids.
2.Assignment Step: Assign each observation to the cluster with the nearest centroid. The distance between a data point and a centroid can be calculated using the Euclidean distance:
$$d(x, \mu ) = \sqrt^ (x_ - \mu _)^}$$
(12)
3.Update Step: Recalculate the centroids for each cluster as the mean of all the data points assigned to that cluster:
$$\mu _ = \frac|} \sum _} x$$
(13)
4.Repeat the assignment and update steps until the centroids no longer change significantly or a predefined number of iterations is reached.
The method will converge to a solution thanks to this iterative process, even if that solution ends up being a local minimum. It is frequently advised to run the algorithm several times using various random initializations and then choose the optimal WCSS solution.
Multiclass classification using One-vs.-The-Rest approachMulticlass classification in machine learning involves categorizing data into more than two classes. The One-vs-The-Rest (OvR) strategy, also known as One-vs-All, is a common method to extend binary classifiers for multiclass problems. This section outlines several classifiers that can be adapted for multi-class classification using the OvR approach, along with their mathematical underpinnings.
The ensemble.Gradient Boosting Classifier uses boosting techniques to optimize a cost function over weak learners, typically decision trees. In the OvR context, it builds a separate model for each class, treating one class as positive and all others as negative. Its objective function is given by:
$$\min _} \sum _^ L(y_, f_(x_)) + }(f_),$$
(14)
where \(f_\) is the model for class i, L is the loss function, and \(x_, y_\) are the features and label of the jth instance.
The Gaussian Process Classifier (GPC) utilizes a Gaussian process (GP) as a prior over functions in a Bayesian framework. For a given dataset, it maps inputs to a latent function space using a kernel (covariance function), typically zero-mean. The kernel defines the similarity between data points, affecting the shape of the function space. Class probabilities are derived by applying a link function, like the sigmoid, to these latent functions. Prediction involves conditioning the GP on the observed data to estimate the posterior distribution, enabling probabilistic classification of new data points.
Linear support vector classification is finding a hyperplane in the feature space that divides the classes with the largest margin is the goal of vector classification. The following optimization problem must be solved as part of the procedure:
$$\min _}, b} \frac \Vert }\Vert ^$$
(15)
subject to
$$y_ (} \cdot }_ + b) \ge 1, \, \forall i.$$
(16)
Here, \(}\) is the weight vector normal to the hyperplane, \(b\) is the bias term, \(}_\) are the feature vectors, and \(y_\) are the class labels (\(+1\) or \(-1\)) for each data point in the training set. The objective of this optimization is to maximize the margin, which is the distance between the hyperplane and the nearest data point from either class. Once the optimal \(}\) and \(b\) are found, classification of a new data point \(}\) is performed using the decision function:
$$}(}) = }(} \cdot } + b).$$
(17)
One statistical technique for binary classification is logistic regression. It simulates the likelihood that an input falls into a specific category. The logistic function, which is defined as follows, forms the basis of the probability prediction.
$$P(y=1|}) = \frac} \cdot } + b)}}.$$
(18)
In this equation, \(}\) represents the feature vector, \(}\) is the weight vector, \(b\) is the bias term, and \(P(y=1|})\) is the probability that the output \(y\) is 1 given the input vector \(}\). The goal of logistic regression is to find the best parameters (\(}\) and \(b\)) that model the relationship between the feature vector and the probability of the output being in a particular class. This is typically achieved through a method like Maximum Likelihood Estimation.
A stochastic Gradient Descent Classifier is a linear classifier that uses stochastic gradient descent for optimization. It updates the model weights for each class iteratively:
$$} \leftarrow } - \eta \nabla L(}; x_, y_),$$
(19)
where \(\eta\) is the learning rate and \(\nabla L\) is the gradient of the loss function.
Each of these classifiers can be effectively used in a One-vs-The-Rest scheme for multiclass classification, allowing them to tackle complex problems involving multiple classes.
Performance assessment metricsIn order to evaluate the effectiveness of classification models, classification metrics are crucial. The most used metrics for classification jobs are shown here, along with their mathematical equivalents.
The easiest performance metric to understand is accuracy, which is just the ratio of properly predicted observations to total observations.
$$Accuracy = \frac,$$
(20)
where \(TP\) is True Positives, \(TN\) is True Negatives, \(FP\) is False Positives, and \(FN\) is False Negatives.
The ratio of accurately anticipated positive observations to all predicted positive observations is known as precision..
$$Precision = \frac.$$
(21)
The ratio of accurately predicted positive observations to all observations made during the actual class is known as recall.
Precision and Recall are weighted averages that make up the F1 Score. As such, this score accounts for both false positives and false negatives.
$$F1\ Score = 2 \times \frac.$$
(23)
A graphical representation called the Receiver Operating Characteristic (ROC) curve shows how well a binary classifier system can diagnose problems as its discrimination threshold is changed. The degree or metric of separability is represented by the Area Under the Curve (AUC).
$$\text = \int _^ \text (t) \, dt.$$
(24)
These metrics offer a thorough understanding of a classification model’s performance by emphasizing various facets of accuracy and error kinds.
Proposed methodThe Clustered-Based Binary Grey Wolf Optimizer (CB-BGWO) is used in the suggested methodology to improve feature selection in high-dimensional genetic datasets. This technique minimizes redundancy and maximizes relevance by combining feature clustering with a binary optimization procedure. In order to evaluate the interdependence of features, Mutual Information (MI) is first calculated. To streamline the search space, characteristics are organized into groups using k-means clustering based on the correlation values. After that, CB-BGWO is performed separately to every cluster, maximizing feature selection by locating the most informative features and reducing redundancy. The final feature set is created by combining the chosen features from each cluster. The performance is then assessed using metrics like accuracy, F1-score, recall, and precision after this optimized set is utilized for classification. The process is divided into four distinct phases:
Step 1: mutual information (MI) computation* MI is a powerful statistical tool that quantifies the mutual dependence between variables. In our context, it helps in understanding how closely related different genetic features are with respect to NHL and various rheumatologic diseases.
Process we compute the MI between pairs of features to evaluate their correlation. A higher MI value indicates a stronger relationship, suggesting a significant role in disease manifestation or progression.
Importance this step is crucial for identifying key genetic markers that could be instrumental in understanding the pathogenesis of these diseases. By focusing on correlated features, we can streamline our analysis to the most relevant genetic factors.
Step 2: feature clusteringThe clustering of features based on similarity followed by a rigorous selection process ensures that only the most relevant and non-redundant features are considered for further analysis.
Step 3: apply binary grey wolf optimizer (bGWO) on each clusterThis optimization algorithm is utilized to sift through the clustered features, selecting those that offer the highest relevance to our study. The bGWO is known for its efficiency in handling complex feature spaces, making it ideal for our study.
Step 4: final feature set identificationThis phase is about distilling our feature set to the most impactful and informative markers, which are likely to yield the most significant insights when used for classification.
Aggregation the features that were chosen through BGWO (Binary Grey Wolf Optimization) are methodically combined in each cluster, resulting in the consolidation of an extensive collection of attributes.
Step 5: classificationThe final phase involves applying a classification method to the selected features to validate their predictive power and practical relevance in diagnosing or understanding NHL and associated rheumatologic diseases.
In conclusion, each phase of our methodology is designed with a clear purpose and is backed by logical reasoning. This structured approach ensures a comprehensive and effective analysis of the complex genetic interactions at play in NHL and rheumatologic diseases.
Our algorithm’s implementation of the novel Clustered-Based Binary Grey Wolf Optimizer (CB-BGWO) is a key component (Algorithm 1). By applying this optimizer to every cluster, we can improve the feature selection procedure and maximize the identification of important genetic variables for our investigation. The algorithm then gathers the final set of features from every cluster and uses this improved feature set to apply a classification technique. Lastly, it assesses the model using common metrics like F1-score, recall, accuracy, and precision.
Figure 3 displays a flowchart that visually represents the specific steps of our algorithm. This flowchart helps to clarify the methodological workflow and improve comprehension of the steps and organization of our approach.
Algorithm 1
Clustered-based Binary Grey Wolf Optimizer (CB-BGWO) Methodology
Fig. 3
Proposed framework pipeline for feature selection and classification using CB-BGWO
Comments (0)