
Remember me

Ethics committee approval was obtained from the London—Dulwich research ethics committee (REC no: 18/LO/0190). This study protocol adhered to the Declaration of Helsinki. Eligible patients were recruited in clinic and each patient voluntarily gave written informed consent to participate in the study. The study is registered at clinicaltrial.gov (NCT03210571).
Inclusion and exclusion criteriaPatients aged 18 years or older with refractory primary open angle (POAG), pseudoexfoliative (PXF) or pigmentary (PG) glaucomas after previous failed filtration surgeries and who were candidates for a GDD were consecutively included. Patients with the diagnosis of angle closure glaucoma, secondary glaucomas (e.g. neovascular, uveitic, silicone oil, congenital), angle dysgenesis, microphthalmia, optic neuropathy secondary to other causes than glaucoma, diabetic retinopathy graded more than severe non-proliferative, previous strabismus or corneal transplantation surgery, previous retinal detachment or pars plana vitrectomy surgery and history of ocular trauma were all excluded. Patients unable to complete the required post-operative visits were also excluded.
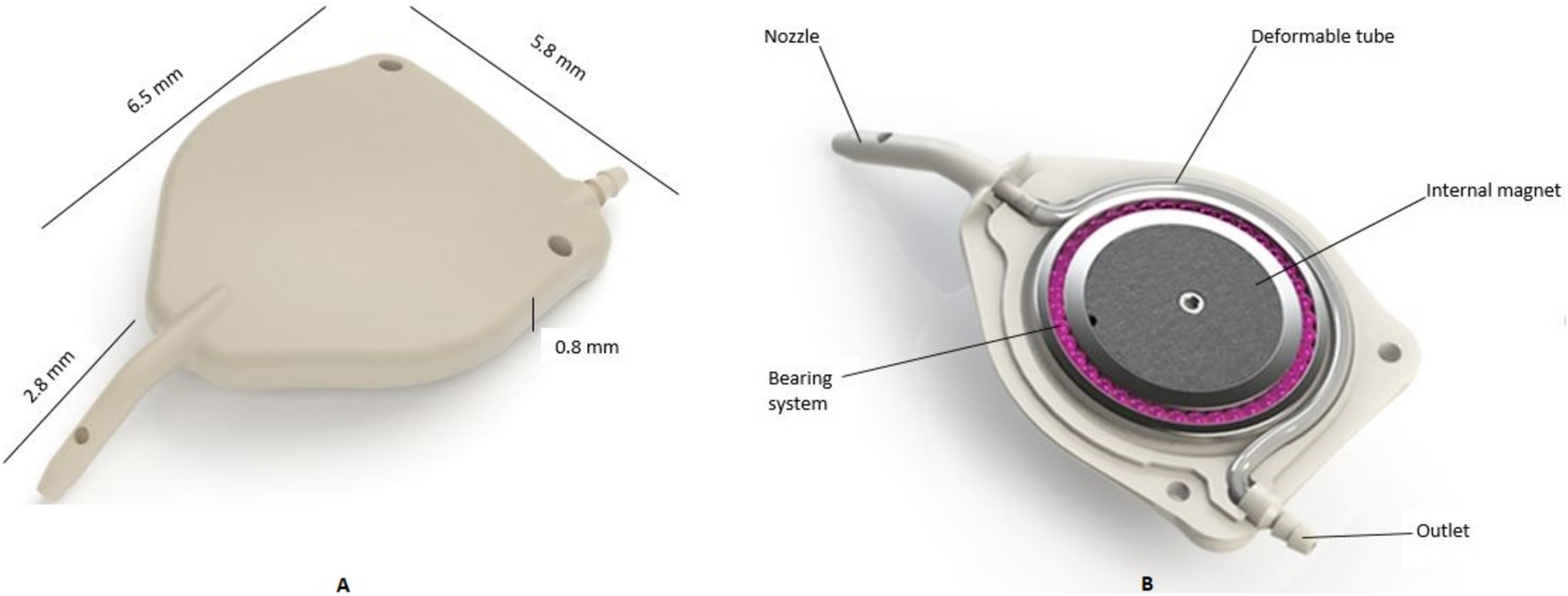
Surgical techniqueAll surgeries were performed by site investigators (LA or KSL) under local or general anaesthesia. Superior temporal placement was the preferred position of the eW (Fig. 4).
Fig. 4
Implantation of the eyeWatch™ system
A fornix-based peritomy was made at the superior temporal limbus, along with exposure of the sclera by careful dissection of the Tenon’s capsule. Gentle wet-field cautery was used to achieve haemostasis. The eyePlate was placed 10–12 mm from the limbus, between the rectus muscles and secured with two single Prolene 7–0 sutures. A 25-gauge needle was used to fashion a track, enabling the device’s nozzle to be inserted into the anterior chamber. A superficial sclerectomy was performed using the outline of the eW device 2 mm from the limbus, enabling the device to be fixed securely using two single Nylon 9–0 sutures. The eW was initially delivered on the opened position (position 0). The magnet was then closed (position 6) in order to increase the fluidic resistance. The eyePlate tube was trimmed appropriately and fixed to the eW device. A pericardium patch (Tutoplast, Tutogen Medical GmbH) of about 6 × 6 mm was finally secured on the eW device using interrupted Nylon 10–0 sutures, with conjunctival closure using interrupted Vicryl 8–0 sutures. Patients administered a post-operative treatment regime consisting of topical antibiotics (Chloramphenicol 0.5% preservative free, Bausch & Lomb, UK) four times a day for two weeks and corticosteroids (Dexamethasone 0.1% preservative free, Rayner Pharmaceuticals, UK) every two hours for the first month; tapered in a reducing dose over the next two months.
Follow-up and study measurementsFollow-up visits were performed at day 1, week 1, week 2, month 1, month 2, month 3, month 6 and month 12 after surgery. Best-corrected distance visual acuity (BCVA) was assessed using Logarithm of the Minimum Angle of Resolution (logMAR). Pachymetry was performed using a non-contact pachymeter (Pachmate 2; DGH Technology, Pennsylvania, USA) and corneal endothelial cell density using specular microscopy (EM 3000, Tomey, Arizona, USA). All patients underwent slit lamp biomicroscopy, Goldmann applanation tonometry, gonioscopy and optic disc evaluation where appropriate.
Goldmann tonometry and reading of the eW position was performed by the site investigator who performed the procedure (LA or KSL). The eW implant was adjusted in one of two ways: if IOP was raised above 16 mmHg, the rotor was adjusted to decrease outflow resistance. Alternatively, if the IOP was low (i.e. < 6 mmHg), the rotor was adjusted to increase outflow resistance and increase IOP to the desired value. The method of adjustment of the rotor with the eyeWatch™ Pen (Fig. 5) has previously been described [12].
Fig. 5
Adjustment of the eyeWatch™ system: a—positional outflow measured using the compass of the eyeWatch Pen. b—eyeWatch™ Pen turned around to reveal external magnet. c—EyeWatch™ Pen turned in direction desired. d—New position of the eyeWatch™ implant confirmed with the eyeWatch™ Pen compass
Outcome measuresThe outcome measures used in this study were similar to those in previous studies evaluating the implant connected with BGI plate [11, 13]11 13 and are summarised below:
1.The primary outcome measures obtained were IOP, BCVA, glaucoma topical medications and number of complications.
2.The primary endpoint was the ability of the eyePlate combined to an eW implant to reduce IOP by ≥ 20% or IOP < 21 mmHg, with no IOP < 6 mmHg on two consecutive visits after 12 months. A complete success was classified as an IOP ≤ 18 and ≥ 6 mmHg with a reduction by 20% from baseline at the last follow-up visit, without the need for glaucoma medications. Overall success was classified as an IOP ≤ 21 and ≥ 6 mmHg and a reduction by 20% from baseline at the last follow-up visit with the use of glaucoma medications.
3.Failure was defined as need for additional glaucoma surgical intervention, loss of light perception, explanting of device, IOP > 21 mmHg or < 20% reduction from baseline on 2 visits after 3 months or persistent hypotony (IOP < 6 mmHg on 2 consecutive study visits after 3 months). Additional glaucoma surgical intervention was defined as the requirement for a further filtering procedure or revision to be performed in theatre. Slit lamp interventions e.g. paracentesis or reformation of anterior chamber were not considered to be in this category.
Statistical analysisData were processed as mean ± SD or as percentages where appropriate. Normality of data was assessed by Shapiro–Wilk test. Comparison of pre and post-operative data was processed via paired t-test for parametric and Wilcoxon signed-rank test for non-parametric distributions. All statistical analyses were performed using Statistical Package for Social Sciences (SPSS) version 26 (IBM Corp, Chicago, USA). A p-value of ≤ 0.05 was considered statistically significant.
Comments (0)