
Remember me

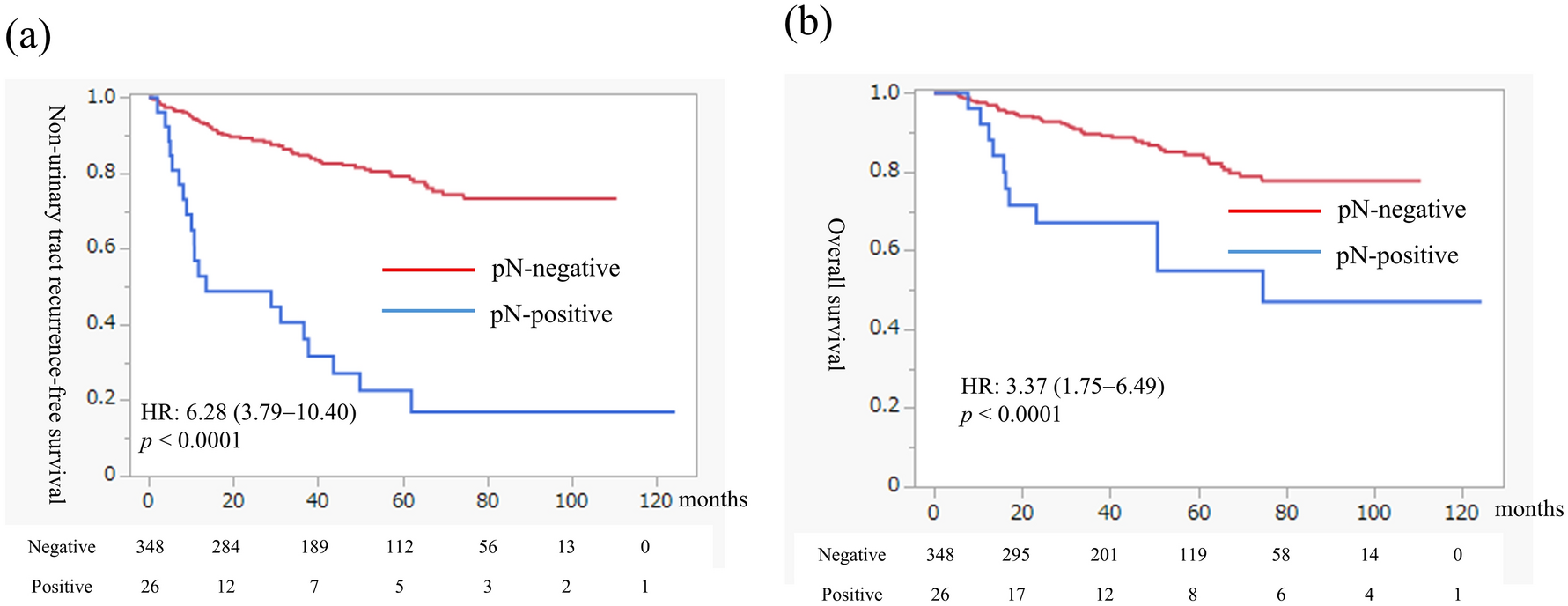
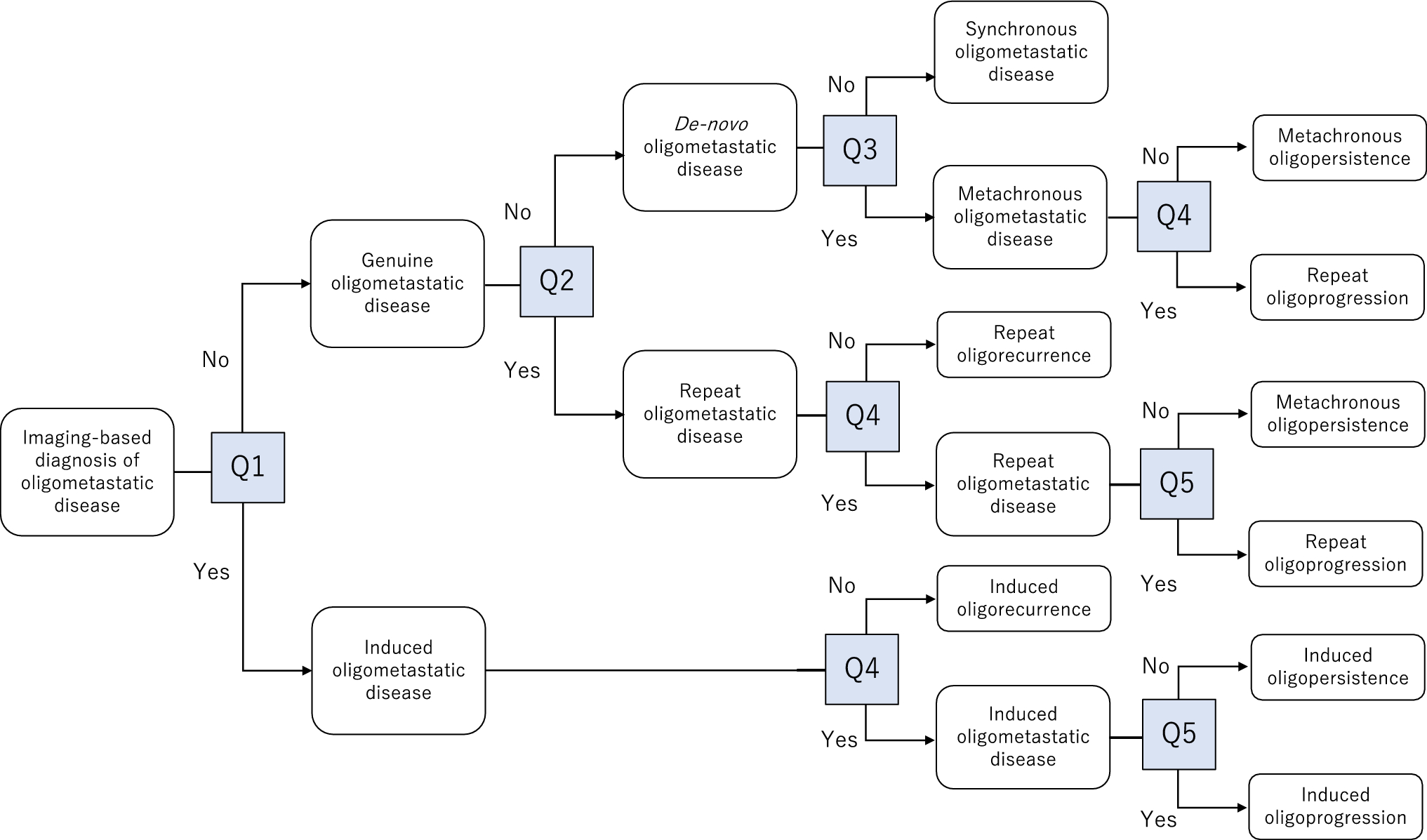
This was a Phase 1, open-label, non-randomized, dose-escalation evaluation of tusamitamab ravtansine administered intravenously as monotherapy (Q2 W or Q3 W) in Japanese adults with advanced solid tumors (NCT03324113). The study comprised three different dose-escalation parts: (i) a main dose-escalation part with Q2 W administration of tusamitamab ravtansine (Fig. 1a); (ii) an LD part with Q2 W administration of tusamitamab ravtansine with a LD at C1, followed by 100 mg/m2 Q2 W (Fig. 1a); and (iii) a dose-escalation Q3 W part with Q3 W administration of tusamitamab ravtansine (Fig. 1b). The decision to escalate to the next dose level (DL) was based on the results of the FIH Phase 1 study (NCT02187848), and the dose-escalation decision rule was defined using a modified toxicity probability interval-2 (mTPI-2) approach.
Fig. 1
A Treatment schema for dose-escalation part (main and bis with loading dose); B Treatment schema for the dose-escalation Q3 W part. C cycle; D day; DLT dose limiting toxicity; IC informed consent; PD progressive disease; Q2 W every two weeks; Q3 W every three weeks; R registration
The study was conducted in accordance with the Declaration of Helsinki, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, and Good Clinical Practice guidelines. The protocol and all amendments were approved by the Ethics Committee or Institutional Review Board at each study site. All patients provided written informed consent before participating in the trial.
Eligibility criteriaInclusion criteriaEligible Japanese patients (aged ≥ 20 years) had CEACAM5-expressing locally advanced or metastatic solid malignant tumors. CEACAM5 expression was assessed retrospectively using immunohistochemistry on the most recent formalin-fixed paraffin-embedded archival tissue samples. Circulating CEACAM5 expression levels were used for enrichment.
Exclusion criteriaPatients with an Eastern Cooperative Oncology Group (ECOG) performance status of ≥ 2, a life expectancy of < 12 weeks, and a prior therapy targeting CEACAM5 or prior maytansinoid treatment were excluded from the study. Detailed inclusion and exclusion criteria are mentioned in Supplementary Table 1.
Study interventionsTusamitamab ravtansine was administered intravenously on Day (D) 1 and repeated Q2 W for the main dose-escalation and dose-escalation parts with LD or repeated Q3 W for the dose-escalation Q3 W part, until toxicity, disease progression, or any other discontinuation criteria (consent withdrawal, unacceptable adverse events [AEs], poor compliance to study protocol, or patient lost to follow-up). Intra-patient dose-escalation and re-escalation were not allowed for any part.
Tusamitamab ravtansine was administered intravenously at the starting dose of 80 mg/m2 in main dose-escalation Q2 W part; additional DLs assessed are mentioned in Table 1. For dose-escalation bis part with LD at Cycle 1 followed by 100 mg/m2 Q2 W, the investigation of LD levels with mTPI-2 design was planned for 120 mg/m2, 135 mg/m2, 150 mg/m2, 170 mg/m2, and 190 mg/m2 (Table 1), followed by treatment with the MTD as 100 mg/m2 Q2 W cycle. The starting level of LD was 135 mg/m2; doses up to 170 mg/m2 were investigated. Two of the three DLT-evaluable patients at the 190 mg/m2 DL experienced a DLT in the FIH study. Hence, 190 mg/m2 was not evaluated in this study. Dose-escalation was expected to proceed with 150 mg/m2 as the starting DL in dose-escalation Q3 W part (Table 1).
Objectives and endpointsObjectivesThe primary study objective was to evaluate the tolerability and safety of tusamitamab ravtansine administered as a single agent according to the investigational medicinal product (IMP)-related DLTs to determine the recommended dose of tusamitamab ravtansine in Japanese patients with advanced malignant solid tumors. The key secondary objectives were to characterize the overall safety profile, PK, and pharmacodynamics (PDy) of tusamitamab ravtansine monotherapy. Preliminary efficacy, as per the Response Evaluation Criteria in Solid Tumors (RECIST) 1.1, and the potential immunogenicity of tusamitamab ravtansine were also evaluated. The tertiary objective was to explore the link between tumor-specific CEACAM5 expression and anti-tumor activity of tusamitamab ravtansine monotherapy.
EndpointsThe primary endpoint was the incidence of IMP-related DLTs at C1 and C2 for the main dose-escalation part and dose-escalation bis part with LD, and at C1, for the dose-escalation Q3 W part. The key secondary endpoints included the characterization of adverse events (AEs) or other abnormalities, the assessment of PK parameters after single and repeat doses, the assessment of CEACAM5 expression, and the presence of anti-drug antibodies. Efficacy was documented by tumor response, duration of response, and time-to-progression (TTP). Detailed objectives and endpoints are mentioned in Supplementary Table 2.
AssessmentsSafety was assessed by a physical examination, laboratory test abnormalities, and TEAEs as graded by the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) v4.03. AEs were coded to a Preferred Term (PT) and the associated primary System Organ Class (SOC) using the Medical Dictionary for Regulatory Activities (MedDRA 25.1), and summarized by type, frequency, severity, seriousness, and relatedness. Tusamitamab ravtansine concentrations in plasma were determined using a validated immunoassay detecting the ADC bearing at least one DM4 molecule. PK parameters at C1 were calculated by a standard non-compartmental analysis. Tumor response, documented as complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD), was assessed using RECIST v1.1 if patients had measurable disease at baseline.
Statistical analysisAll analyses were descriptive unless otherwise specified and performed based on the all-treated population. DLT-evaluable patients had completed two cycles and received at least 80% of the intended dose at each of the first two infusions unless they discontinued the IMP before the completion of C2 due to a DLT. In the dose-escalation Q3 W cohort, all treated population had completed C1 and received at least 80% of the intended dose at the first infusion unless they discontinued the IMP before the end of C1 due to a DLT. Continuous data were summarized using the number of available data, mean, standard deviation, median, minimum, and maximum for each DL. Categorical and ordinal data were summarized using the number and percentage of patients in each DL.
Comments (0)