As William Osler astutely remarked, “to study the phenomena of disease without books is to sail an uncharted sea, while to study books without patients is not to go to sea at all.” This axiom underscores the imperative integration of theoretical and clinical perspectives in medicine. Mounier–Kuhn's seminal 1932 report revolutionized our understanding of this condition through pioneering endoscopic and radiographic investigations, ultimately leading to its eponymous designation as MKS [3]. Comprehensive analysis of MKS thus requires dual examination of its clinical spectrum and historical diagnostic evolution, with bibliometric data demonstrating sustained global research productivity despite annual publication fluctuations.
MKS manifests as idiopathic tracheobronchial dilation with characteristic histopathological findings: atrophy of airway wall elastic fibres and smooth muscle layer hypoplasia [8]. Progressive parenchymal deterioration represents a hallmark pathophysiological feature of this disease [9], with symptom onset spanning 18 months to 84 years of age (peak incidence: 30–50 years). In our cohort, the mean age at diagnosis was 55.9 ± 16.6 years, consistent with previously reported epidemiological trends. Our findings confirm a striking male predominance (male-to-female ratio 5.5:1), aligning with epidemiological reports of ≈8:1 male-to-female disparities [10]. The etiopathogenesis remains debated, with two prevailing theories: (1) congenital origin via autosomal recessive inheritance, supported by autopsy evidence of tracheobronchial submucosal tissue deficiency [11]; and (2) acquired pathogenesis from prolonged mechanical ventilation-induced airway remodelling, particularly in premature infants [12]. Notably, our cases showed no familial clustering or comorbidities with connective tissue disorders (Ehlers–Danlos syndrome, Marfan syndrome), which are occasionally associated with MKS [13].
MKS exhibits marked interindividual variability in clinical presentation, with nonspecific respiratory manifestations predominating. Chronic cough (64.5%), productive sputum (44.4%), and progressive dyspnoea (52.7%) constitute the cardinal triad, whereas fever (22.5%) and haemoptysis (14.2%) represent less frequent features. Symptom onset typically emerges in the third decade,although paediatric-onset recurrent infections are documented. Notably, 8.3% of patients remain asymptomatic at initial diagnosis, although few maintain a lifelong symptom-free status. Frequent comorbid manifestations include bronchiectasis (71.6%) and recurrent pneumonia (57.4%). Emerging evidence suggests potential SARS-CoV-2-associated tracheomegaly progression, exemplified by a documented case of post-COVID-19 symptomatic exacerbation [14]. Extrathoracic associations encompass nasal polyposis and congenital craniofacial anomalies—bilateral ptosis, epicanthal folds, micrognathia, and upper lip redundancy—observed in syndromic variants [15]. Laryngeal involvement, manifesting as progressive hoarseness secondary to vocal cord paralysis or cricoarytenoid joint remodelling, has been mechanistically linked to tracheobronchial wall instability [16].
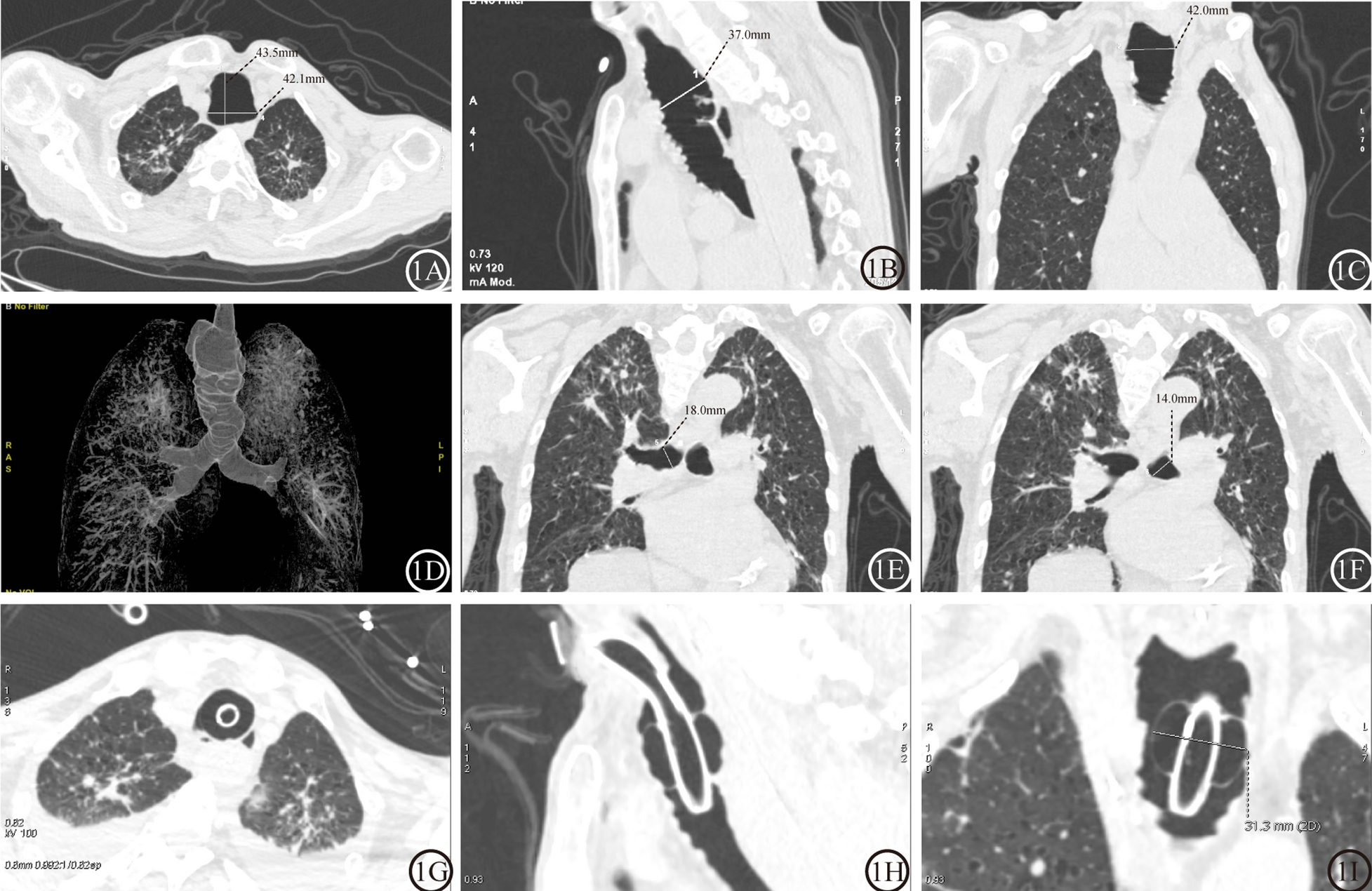
Typically, tracheal diameters exceeding sex-specific thresholds (coronal: males ≥ 25 mm, females ≥ 21 mm) remain pathognomonic [6, 17]. Posteroanterior chest radiography may reveal tracheal luminal expansion approximating the vertebral body width, which is more conspicuous in lateral projections. High-resolution computed tomography (HRCT) constitutes the diagnostic gold standard (see Methods) [18]. Three-dimensional CT reconstructions demonstrate pantracheobronchial dilation with characteristic saccular protrusions between cartilage rings, correlating bronchoscopically with dynamic mucosal herniation during forced expiration [19,20,21]. Alterations in the fluorescence of fibres have also been observed via confocal microscopy in patients with tracheobronchomegaly syndrome [8]. Autofluorescence imaging (AFI), when integrated with bronchoscopy, provides real-time visualization of tracheobronchial mucosal changes. Diminished AFI signal intensity in these regions correlates histopathologically with elastic fibre depletion or atrophy [22].
In summary, an MKS diagnosis hinges on the integration of characteristic clinical profiles and pathognomonic imaging criteria. The median diagnostic delay of 3.0 years (IQR: 0.25–20.0 years) and extreme interquartile variability in MKS suggest persistent underdiagnosis. Our case-driven insights position MKS as a critical differential in patients exhibiting chronic cough, sputum production, and haemoptysis alongside refractory lower respiratory infections. Severe manifestations may progress to exertional dyspnoea complicated by bronchiectasis-related haemoptysis [23], with rare presentations including spontaneous pneumothorax, life-threatening haemoptysis, and digital clubbing [24]. When conventional therapies fail in this context, multidisciplinary evaluation should prioritize dynamic airway assessment via forced-expiration CT or bronchoscopy to confirm the tracheobronchomalacia patterns. Standardized educational protocols integrating early HRCT evaluation of central airway dilation and recurrent pneumonia etiologies may mitigate diagnostic delays, especially in subclinical presentations.
The diagnostic workup must rigorously differentiate MKS from acquired tracheobronchial dilation and structural variants. Key exclusions include the following: (1) fibrotic tracheomegaly secondary to pulmonary fibrosis-induced opposing traction; (2) mucosal pseudodilation syndromes (laryngoceles, Zenker's diverticulum) lacking true airway wall pathology; and (3) apical lung herniations and bullous emphysema (airspace > 1 cm without tracheal involvement). Williams-Campbell syndrome is characterized by congenital cystic bronchiectasis resulting from a lack of cartilage in the fourth- to sixth-order bronchi [4, 25,26,27,28,29,30].
Postdiagnosis, a comprehensive evaluation of respiratory functional impairment grading and disease trajectory is critical for prognostication. Pulmonary function tests may reveal obstructive patterns with elevated residual volumes or they may remain normal [25]. In our cohort, spirometry demonstrated moderate obstruction in one patient and mild restriction in two other patients. Innovative assessment protocols, such as Pacheco’s single-breath nitrogen washout method, address MKS-specific challenges by quantifying anatomical dead space [9]. The nosological classification of MKS remains contentious, with two prevailing systems (Table 3). The Himalstein classification (1973) stratifies MKS into three types according to tracheobronchial dilation severity [31], whereas Payandeh’s framework (2015) derives from a meta-analysis of 365 MKS cases across 166 studies, emphasizing its etiopathogenetic heterogeneity [10]. While the Himalstein system emphasizes structural abnormalities, the Payandeh framework incorporates etiopathogenetic considerations. This dichotomy underscores the necessity for clinicians to integrate multimodal clinical–radiological data for precise phenotyping.
Table 3 Comparative classification systems for MKSOur study reveals that conservative management remains the cornerstone of MKS therapy, employed in 70.4% of cases (119/169). This aligns with current guidelines emphasizing symptom control through airway clearance and infection prevention in this anatomically driven disorder [11]. However, the modest rate of clinical stabilization (62.7%) and equivalent proportions of symptomatic improvement and treatment failure (8.3% each) underscore the limited therapeutic efficacy of conventional approaches. For asymptomatic MKS patients, therapeutic algorithms focus on infection prophylaxis, risk factor modulation, smoking cessation, and occupational irritant avoidance. During acute exacerbations with hypersecretion, strategies prioritize augmenting mucociliary clearance mechanisms via mucolytic agents and chest physiotherapy modalities (e.g., postural drainage). Targeted antibiotic regimens address superimposed infections guided by microbiological data [32].
Despite optimized management, progressive respiratory insufficiency may ensue, necessitating invasive interventions such as airway stenting or tracheobronchoplasty for severe dynamic collapse—although limited experience in MKS cohorts shows variable efficacy and frequent complications (infection, stent migration) [1, 33, 34]. Lung transplantation is reserved for patients whose end-stage respiratory failure is refractory to conventional therapies [35,36,37]. Notably, endoscopic laser ablation achieved sustained symptom remission in a 68-year-old MKS patient, demonstrating novel therapeutic potential [38]. The low utilization of ventilator-assisted support (11.2%) and surgery (10.1%) may reflect either under-recognition of advanced disease stages or reluctance to escalate care in a condition with no curative options. Notably, the 1:1 ratio between improvement and failure signals critical heterogeneity in treatment response, potentially tied to variations in tracheobronchial dilation severity or comorbid burden.
Perioperative airway management in MKS requires meticulous multidisciplinary planning. Surgical anaesthesia or prolonged positive-pressure ventilation mandates comprehensive otolaryngological–anaesthesiological evaluation [39]. Subglottic cuff placement with leak-controlled inflation optimizes tidal volume maintenance during intubation, which is complemented by laryngeal mask airways or oropharyngeal packing to prevent air leakage [40,41,42]. Modified laryngotracheal separation techniques enable high-pressure ventilation in neonates with tracheobronchomegaly and severe bronchopulmonary dysplasia [43]. Postprocedural monitoring of cuff pressures remains critical to balance tracheal seal efficacy and wall integrity preservation, as illustrated by Patient 1’s air leakage complications [44]. Therapeutic decisions require algorithm-driven selection on the basis of disease phenotype, pulmonary reserve, mechanical ventilation needs, and airway stability parameters.




Comments (0)