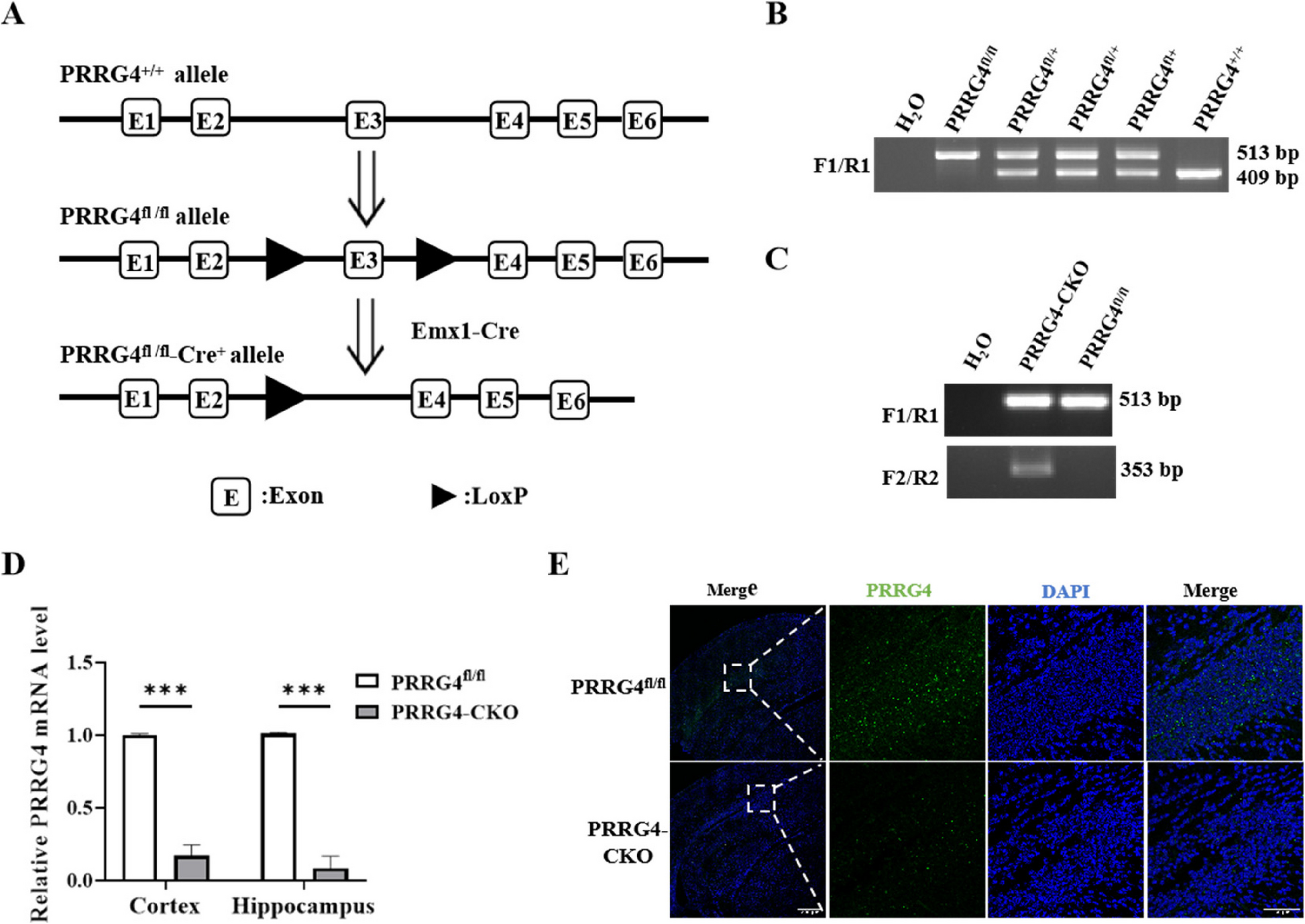
Generation of PRRG4 Brain-Specific Conditional Knockout Mice and Housing of the Experimental Mice
PRRG4fl/+ mice with a C57BL/6 background with LoxP sites flanking exon 3 of PRRG4 were generated by GemPharmatech Co., Ltd (Nanjing, China) using CRISPR-Cas9 technology. Emx1-Cre mice (C57BL/6) were kindly provided by Dr. Jieguang Chen at Wenzhou Medical University. In this study, the Emx1 promoter was used to drive Cre expression in the cerebral cortex and hippocampus. Studies have reported that the Emx1 promoter specifically drives expression in the cerebral cortex and hippocampus of mice [12]. PRRG4fl/fl mice were obtained by mating PRRG4fl/+ mice with each other. PRRG4fl/+-Cre+ mice were obtained by mating Emx1-Cre mice (i.e., Cre+ mice) with PRRG4fl/fl mice. PRRG4fl/fl-Cre+ (PRRG4 brain-specific conditional knockout, referred to as PRRG4-CKO) mice and normal control PRRG4fl/fl mice were obtained by mating PRRG4fl/+-Cre+ mice with PRRG4fl/fl mice. Mice were housed under 12 h light/dark cycle, temperature 20℃−25℃, humidity 40%−60%, with sufficient water and food. At the end of the experiment, all mice were euthanized using CO2 followed by cervical dislocation. All animal experiments were approved by the Animal Care and Use Committee and Ethics Committee of Wenzhou Medical University (wydw2023-0214).
PCR Genotyping of Mice
Tail specimens were clipped from mice at 7–10 days after birth, DNA was extracted, and genotypes were identified by conventional PCR. To identify whether the PRRG4 gene carries LoxP sites, a pair of primers F1/R1 was designed upstream and downstream of the 5'LoxP site. To identify Cre+ mice, in addition to F1/R1 primers, a pair of primers F2/R2 located upstream and downstream of the Cre gene was designed. Primer sequences from 5' to 3': F1: CTGCCAGGAAAGAAGAGTAGGACA, R1: TTACCCATTCAGCTGTCTCCCCA, F2: ATGTCCAATTTACTGACCG, R2: CGCCGCATAACCAGTGAAAC.
qPCR Detection of PRRG4 mRNA Expression in Mouse Brain
Mice at postnatal day 10 were anesthetized by inhalation of 1.5% isoflurane, and the cerebral cortex and hippocampus tissues were immersed in an appropriate amount of RNA Keeper Tissue Stabilizer (Vazyme, Nanjing, China) to inactivate endogenous RNase in the tissues. RNA was extracted after homogenization. The extracted RNA was reversely transcribed into cDNA for qPCR. A pair of qPCR primers was designed upstream and downstream of the third exon of PRRG4 mRNA. Reaction conditions: 95℃ 10 s, 55℃ 30 s, 95℃ 5 s, 40 cycles. qPCR primer sequences from 5' to 3': PRRG4-F: CCACTTCTGATCGTACTCAGCC, PRRG4-R: GCGGTGCATAAAGATGTTTGCT, GAPDH-F: TGGCCTTCCGTGTTCCTAC, GAPDH-R: GAGTTGCTGTTGAAGTCGCA.
Immunofluorescence Detection of PRRG4 Protein Level in Mouse Brain
Mice at postnatal day 0 were anesthetized by inhalation of 1.5% isoflurane, perfused with 4% paraformaldehyde solution, and the complete brain tissue was separated and fixed in 4% paraformaldehyde solution at 4℃ overnight. The required parts were cut along the coronal plane with a brain mold, dehydrated with 30% sucrose solution, embedded in OCT embedding agent, snap-frozen in liquid nitrogen, and then transferred to −80℃ for long-term storage. Tissue sections (14 μm) of the cerebral cortex and hippocampus were cut, and after the OCT was completely dried, they were stored at −30℃ for long-term storage. Sections were taken out from −30℃, blocked after antigen retrieval. Primary rabbit anti-PRRG4 antibodies (1:100, cat. no. HPA009040, Sigma, Massachusetts, USA) were added and incubated at 4℃ overnight. Secondary antibody (Alexa Fluor 488 labeled anti-rabbit IgG, Beyotime Biotechnology, Jiangsu, China) was added and incubated at room temperature for 2 h in the dark. Nuclei were stained with DAPI (Beyotime Biotechnology) for 4 min. Anti-fluorescence quenching agent was added to the tissue sections, which were covered by coverslips and imaged under a laser scanning confocal microscope.
Three-Chamber Test for Assessing Social Ability and Social Novelty of Mice
The three-chamber test device was a rectangular box (length × width × height: 60 × 40 × 30 cm), which was divided into three small chambers by partitions, with 5 × 5 cm openings in the partitions. A mouse restraint cage (diameter 8 cm, height 10 cm) was placed in each of the side chambers. The experiment was divided into three stages: habituation stage, social ability test stage, and social novelty test stage. In the habituation stage, both sides were empty cage E, and experimental mice (2–3 months old, 10–11 mice/group) were placed in the middle chamber and allowed to freely explore the two empty cages for 10 min. In the social ability test stage, a non-littermate wild-type mouse (Stranger1, S1) was placed in a cage on one side, and the other side was still an empty cage E. The experimental mice were allowed to freely explore the cage with the stranger mouse S1 and the empty cage E for 10 min. In the social novelty test stage, another non-littermate wild-type mouse (Stranger2, S2) was placed in the other side cage. The experimental mice were allowed to freely explore the cage with the familiar mouse S1 and the cage with the stranger mouse S2 for 10 min. The movement trajectories of all experimental mice were recorded by a video system, and the time spent by the experimental mice in different chambers and cages during different stages was analyzed using behavioral analysis software. Sniffing, direct contact, or climbing the cage by the mice were all counted as cage contact time.
Marble Burying Test and Repetitive Behavior Test for Assessing Stereotypic and Repetitive Behavior in Mice
Before the marble burying test, clean corn cob (5 cm thick) bedding was spread evenly in the cage (45 × 30 × 25 cm). Different colored glass marbles (~ 1.6 cm) were randomly placed on top of the bedding. Photos were taken to record the original state of the marbles. Then, 2–3 months old experimental mice (10–11 mice/group) were placed in the cage for 30 min. Photos were taken to record the marble burying by the mice. The number of buried marbles was counted when more than 2/3 of the marble surface area was covered by the bedding.
The repetitive behavior test was to observe the spontaneous stereotypic and repetitive behavior of mice in a familiar environment. Experimental mice (2–3 months old, 10–11 mice/group) were placed in their daily living cages and allowed to acclimate for 15 min. Videos were recorded to assess the repetitive grooming by the experimental mice within 10 min. Grooming behaviors included grasping the face, head, or body with two forelimbs, or licking body parts.
Elevated Cross Maze Test and Open Field Test for Assessing Anxiety Behavior in Mice
The elevated cross maze device was 60 cm above the ground. The horizontal and vertical lengths of the cross-shaped maze were both 50 cm, representing the open and closed arms, respectively. At the beginning of the experiment, experimental mice (2–3 months old, 10–11 mice/group) were placed into the maze from one side of the closed arm, facing away. The test time was 5 min. The movement of the mice in the maze, and the time and number of entries into different arms were recorded. Finally, the anxiety of the mice was assessed by analyzing the time and number of entries into the open arms using behavioral analysis software.
The open field test was performed in a 40 × 40 × 40 cm experimental box. Experimental mice (2–3 month olds, 10–11 mice/group) were placed in the center of the box, and their movement trajectories were recorded by video for 10 min. Behavioral analysis software was used to analyze the movement distance of the mice to assess their motor ability, and the time spent by the mice in the central area of the open field was mainly analyzed to assess their anxiety status.
Golgi Staining to Detect Dendritic Morphology of Pyramidal Neurons in Mouse Brain
After anesthetizing mice (3 months old, 3 male mice/group) with 1.5% isoflurane inhalation, complete brain tissue was dissected. A Golgi staining kit (FD Neurotech, Columbia, MD, USA) was used according to the manufacturer’s instructions. The brain tissue was immersed in a mixture of solution A and solution B (1:1) and placed at room temperature in the dark for two weeks. It was then transferred to solution C and placed at room temperature in the dark for 3 days, and placed at −80℃ for at least 1 h. Brain tissue was taken out from −80℃, and 100 µm cerebral cortex and hippocampal sections were cut and air-dried naturally at room temperature in the dark. The sections were placed in a mixture of solution D, solution E, and ddH2O (1:1:2) for 10 min for alkalization. After dehydrated with ethanol and permeated with xylene, the sections were covered with coverslips. Microscopic images were taken.
The Neuroanatomy-SNT plugin in Image J software was used to visualize neurons, track neuronal dendrites, and measure the total dendritic length of each pyramidal neuron. The Neuroanatomy-Sholl plugin in Image J software was used to analyze the visualized neurons. Concentric circles with a 10 μm radial increment were drawn with the cell body as the origin. The number of intersections of each pyramidal neuron dendrite on all concentric circles was used as the result of dendritic branching. Image J software was used to count the number of spines on the dendrites, and the Segmented line plugin in Image J software was used to measure the dendritic length. The number of spines per 10 μm dendritic length was used as the dendritic spine density.
Immunoblotting
After anesthetizing mice (6 male mice/group, 3 months old) with 1.5% isoflurane inhalation, complete fresh brain tissue was dissected, and the cerebral cortex and hippocampal tissues were further separated. Brain tissues were homogenized in RIPA buffer (Beyotime Biotechnology), lysed at 4℃ for 30 min, and centrifuged at 12,000 g for 20 min. The supernatant lysates were analyzed by the BCA method, separated by SDS-PAGE electrophoresis, transferred to PVDF membrane, and immunoblotted with corresponding primary antibodies including anti-PSD95 antibody (1:2000; cat. no. 3450; Cell Signaling Technology), anti-SYP antibody (1:8000; cat. no. A6344; ABclonal Technology, Wuhan, China), anti-MAGI2 antibody (1:5000; cat. no. 25189–1-AP; Proteintech, Wuhan, China), anti-RhoA antibody (1:1000; cat. no. SAB4501661; Sigma), anti-β-actin antibody (1:2000; cat. no. 3700; Cell Signaling Technology), and incubated at 4℃ overnight. Secondary antibodies were added, including HRP-labeled anti-mouse antibody (1:5000; cat. no. A0216; Beyotime Biotechnology) or HRP-labeled anti-rabbit antibody (1:5000; cat. no. A0239; Beyotime Biotechnology), and incubated at room temperature for 1 h. The immunoblots were developed using ECL reagent. Band densitometry analysis was performed using Image Lab software.
Generation of SH-SY5Y Cells Overexpressing PRRG4
HEK293T17 cells were co-transfected with recombinant lentiviral plasmid pCDH-PRRG4-Flag, or empty vector plasmid pCDH together with the packaging plasmid pSPAX2, and pMD2G. Lentiviral supernatants were harvested 48 h post transfection, and used to infect human neuroblastoma cell line SH-SY5Y purchased from ATCC in the presence of 8 μg/mL polybrene. The medium was changed after 16 h of infection, and 1.0 μg/mL puromycin was added to the culture dish 6–8 h later.
Preparation and Purification of His-PRRG4 Antigen Protein
IPTG was used to induce host bacteria to express His-tagged mouse PRRG4 antigen protein (aa135-226) by the PET-28a vector: Bacteria culture was induced with IPTG (final concentration 1 mmol/L) at 37℃ for 4 h, and centrifuged at 4℃, 4000 rpm for 10 min. The bacteria pellets were fully resuspended in denaturing lysis buffer containing 8 M urea and 2 mM PMSF and broken by ultrasonic disruption. The resin of the nickel column was mixed with the denatured PRRG4 protein and incubated on a 4℃ rotary shaker overnight. The resin was repeatedly washed with denaturing lysis buffer and eluted with denaturing elution buffer. The eluted denatured PRRG4 protein was renatured by dialysis using PBS.
Purification of Rabbit Anti-Mouse PRRG4 Polyclonal Antibody
To prepare the PRRG4 affinity column, purified PRRG4 antigen (~ 1 mg) was covalently crosslinked to the SulfoLink™ Coupling Resin (Thermo Fisher, Massachusetts, USA) according to the manufacturer’s instruction. Rabbit sera from rabbits immunized with His-tagged mouse PRRG4 protein (aa135-226) were generated by HUABIO (Hangzhou, China). PRRG4 serum (2 ml) was added to the PRRG4 affinity column and incubated with rotation at 4℃ overnight. The bound PRRG4 antibodies were washed and eluted with 100 mM glycine, pH 2.5.
Co-Immunoprecipitation
Cell samples were lysed with lysis buffer containing 50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 20 mM beta-glycerol, 10 mM NaF, 2 mM Na3VO4, and protease inhibitor cocktail (Bimake, Houston, USA). Fresh cerebral cortex and hippocampal tissue samples from mice (3 male mice/group, 3 months old) were homogenized and lysed with the lysis buffer at 4℃ with rotation for 1 h, and clarified by centrifugation. Purified PRRG4 antibodies (or negative control rabbit antibody IgG) were added at 0.6 µg antibody/1 × 106 cell lysate or 1.0 µg antibody/1 mg tissue lysate and incubated for 2 h, followed by addition of 10 µL protein A agarose bead (50% slurry) and overnight at 4℃. Then, protein A agarose beads were washed with lysis buffer for 5 times, boiled with 1 × SDS loading buffer at 95℃ for 5 min, and detected by immunoblotting with PRRG4 and MAGI2 antibodies.
GST-RBD Pull-down Assay to Detect Activated RhoA (RhoA-GTP) Content
pGEX-6p-1-GST-RBD plasmid expressing the GST-tagged Rho-binding domain of Rhotekin (GST-RBD) fusion protein was transformed into BL21 host bacteria. The cultured BL21 bacteria were induced with 0.5 mM IPTG for 2 h at 30 °C, pelleted by centrifugation, resuspended in TBS buffer containing 1% Triton X-100, 10 mM DTT, 2 mM PMSF, 300 µg/ml lysozyme, 10 mM MgCl, and 100 µM Dnase I, and sonicated. The broken bacteria lysate was clarified by centrifugation, and was incubated with glutathione (GT) agarose (Ca#16,100, Thermo Fisher) at 4 °C for 90 min. The purified GST-RBD protein on the GT beads were washed with TBS 5 times and stored on ice temporarily. Fresh cerebral cortex and hippocampal tissue samples from mice (3 male mice/group, 3 months old) were homogenized and lysed with buffer B (1% Triton X-100, 0.5% deoxycholate, 200 mM NaCl, 5 mM MgCl2, 10 mM Tris–Cl pH 7.5) (freshly added 2 mM PMSF and 1% protease inhibitors Cocktail) at 4℃ with rotation for 0.5 h. After centrifugation at 4 °C, 12,000 g for 20 min, the supernatant was added to GST-RBD (~ 50 µg) GT beads and incubated at 4 °C with rotation for 1 h. After centrifugation, the GT beads were washed with buffer B five times, and boiled in 100 μL 1 × SDS lysis buffer. GST-PBD pulldowns and input control were analyzed by western blot using RhoA antibody (1:1000, cat. no. SAB4501661, Sigma).
Statistical Analysis
GraphPad Prism 9.0 software was used for statistical analysis. T-test was used to compare the means between two groups, and ANOVA was used to compare the means between two groups in three or more groups. Variance homogeneity was tested first. If the variance was homogeneous, Tukey was used to calculate the P value; if the variance was heterogeneous, Tamhane's T2 was used to calculate the P value. P < 0.05 indicated a significant difference.








Comments (0)