
Remember me







This study was a randomized, controlled, single-blinded, parallel-group trial comparing the effects of low-intensity pulsed ultrasound (LIPUS) and low-level laser therapy (LLLT) on peri-implant marginal bone preservation and soft tissue healing in patients undergoing dental implant surgery. The study was conducted between July 2023 and September 2024.
Ethical considerationsThis trial is reported in accordance with the CONsolidated Standards Of Reporting Trials (CONSORT) statement [31] (Supplementary file 1), This study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by the Research Ethical Committee of the Faculty of Physical Therapy, Cairo University (approval number P.T.REC/012/004647). The study was also registered on ClinicalTrials.gov (NCT05938868) before participant enrollment.
Participants provided written informed consent after receiving comprehensive study information. Participation was voluntary with unrestricted withdrawal rights. Data confidentiality was ensured through secure storage, restricted access, and anonymized publication of results.
SettingsThe study was conducted at a specialized dental clinic in Sheikh Zayed City, Giza, Egypt. This tertiary referral center features advanced equipment including digital radiography, intraoral scanners, and CAD/CAM technology. The facility comprises six dental operatories and dedicated sterilization and consultation areas. Staff includes specialized dental surgeons, periodontists, prosthodontists, and trained assistants, adhering to international infection control and safety standards.
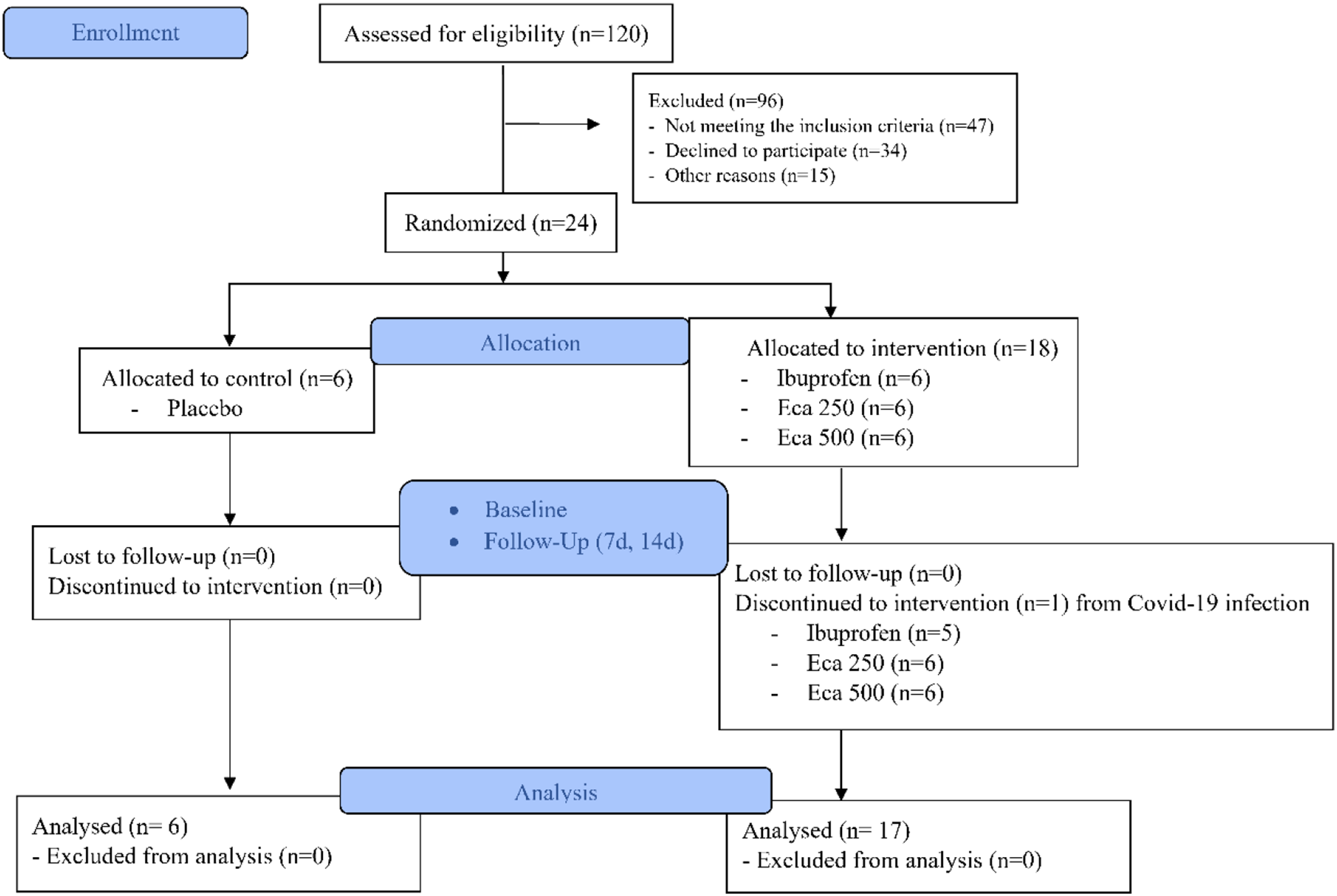
ParticipantsPatient recruitment took place at the outpatient clinics of the Oral-Maxillofacial Surgery and Prosthodontics departments in multiple hospitals across Cairo via healthcare referrals and direct patient contact. Initial screening assessed eligibility per study criteria. Eligible patients received comprehensive study information with opportunities for clarification. The flow of participants through each stage of the trial is illustrated in Fig. 1.
Fig. 1
Flow of participants through the trial
The inclusion criteria were as follows: [1] Adults aged ≥ 18 years requiring dental implants in either the maxilla or mandible; [2] both male and female patients, to allow for gender comparisons; [3] non-smokers, as smoking has been shown to negatively impact implant success rates [32]; and [4] adequate bone volume and density at the implant site, defined as Class III or better according to the Lekholm and Zarb classification [33], as assessed clinically and radiologically.
Study exclusion criteria encompassed: [1] compromised medical status, particularly uncontrolled diabetes; [2] need for bone grafting at the intended implant location; [3] current facial radiotherapy or chemotherapy; [4] suboptimal oral hygiene; [5] previous TMJ disorders; and [6] concurrent oral or periodontal surgical procedures at implant sites.
Sample size calculationThe number of study participants was determined by using G*Power 3.1.9.1; through power analysis based on the following input models; F tests– ANOVA; repeated measures, within-between interaction, given α, power, and effect size. Hence, we assumed 3 groups, 95% confidence interval, 0.05 α level, 80% power, and 0.331 effect size calculated from a previous study which resulted in 54 participants [34]. Considering possible losses, a 15% contingency was added to the number obtained, totaling 63 participants or 21 per group.
Randomization & blindingA randomized, single-blind study design was used to ensure unbiased treatment allocation. Eligible patients were randomly assigned to one of three groups: [1] LIPUS [2], LLLT, or [3] control, using a computer-generated randomization sequence with a 1:1:1 allocation ratio. The randomization sequence was generated by an independent statistician using a permuted block randomization method with a block size of six.
Allocation concealment was maintained using sealed, opaque, sequentially numbered envelopes, opened only after participants completed baseline assessments and provided informed consent. Due to the nature of the interventions, blinding of patients and treating clinicians was not feasible. However, outcome assessors were blinded to the patients’ group allocations.
To maintain blinding, all study-related documents were labeled with unique participant identification numbers rather than group assignments. The study coordinator kept a separate, confidential log linking the identification numbers to the group assignments, which was not accessible to the outcome assessors.
InterventionsAll implant procedures followed an established surgical protocol to ensure procedural consistency [35, 36]. The surgical sequence began with the administration of local anesthetic (2% lidocaine containing 1:100,000 epinephrine). Access to the recipient site was achieved through a midcrestal incision, followed by the reflection of a full-thickness mucoperiosteal flap. Critical anatomical structures were carefully identified and protected throughout the procedure. Implant site preparation was guided by a customized surgical stent. During placement, a minimum clearance of 1.5 mm was maintained between the implant and neighboring dentition to ensure adequate preservation of adjacent hard and soft tissues. The procedure concluded with precise flap repositioning and suturing to obtain tension-free primary closure.
Patients from all three groups received the standard postoperative care. In addition to the standard care, the low-intensity pulsed ultrasound (LIPUS) group received 8 treatment sessions (two sessions per week, 20 min each), while the low-level laser therapy (LLLT) group received 4 treatment sessions (on day 0, 3, 7, and 14 after surgery, 10 s per site). The control group received the standard postoperative care only. Interventions are described according to the template for intervention description and replication (TIDieR) guide [37].
ProceduresLow-intensity pulsed ultrasound (LIPUS) therapyThe LIPUS intervention was delivered using an ENRAF NONIUS, SONOPLUS 492 device (ENRAF NONIUS, The Netherlands, model number: EN-SP492). Patients receiving LIPUS therapy were positioned in a dental chair with their head supported by a headrest. The treatment area was prepared by applying a thin layer of intra-oral gel to the buccal aspect of the implant site to ensure efficient energy transfer. The LIPUS treatment was administered via an intraoral delivery system, utilizing a 0.8 cm2 probe positioned against the buccal surface of the implant region. The probe was placed in direct contact with the buccal attached gingival tissue, separated only by a thin layer of intraoral transmission gel that served as a coupling medium. LIPUS therapy was initiated 48 h post-surgery and administered twice weekly for 4 consecutive weeks, totaling 8 treatment sessions. Each 20-minute session utilized ultrasonic waves at 1 MHz frequency, delivering a power output of 20 mW and maintaining an intensity of 30 mW/cm2 with a static transducer position [38]. Patients were instructed to remain still and relax during the treatment. After each session, the treatment area was cleaned, and the patient was inspected for any complications.
Low-level laser therapy (LLLT)For the LLLT intervention, a Chattanooga device, model 27,841 (Chattanooga, USA), equipped with a gallium-arsenide (GaAs) diode laser was used. The laser device emitted light at a wavelength of 850 nm and operated at a power of 200 mW in continuous wave mode [39].
Patients were seated in a dental chair with their head supported by a headrest. The mucous membrane surrounding the implant site was cleaned and dried before the application of the laser. The laser treatment was administered by applying the probe at 10 mm standoff distance from the soft tissue surrounding the implant, with the beam directed at six distinct points circumferentially around the implant site: mesiobuccally, distobuccal, midbuccal, midlingual, mesiolingual, and distolingual [40]. Each site was irradiated for 10 s, delivering a total energy of 6 J per session. Patients and therapists wore protective goggles during the treatment.
Both devices were calibrated before the start of the study and at regular intervals throughout the trial to ensure consistent output. Calibration was performed using the manufacturers’ recommended protocols and certified equipment.
Standard postoperative care (control group)Patients in the control group received standard postoperative care [19], which included several components:
1.Extra-oral ice pack application: Patients were instructed to apply ice packs on the external surface of the face, over the implant area, for 20 min every 2 h during the first 48 h after surgery.
2.Oral hygiene maintenance: Patients were advised to maintain their daily oral hygiene routine, including gentle brushing and rinsing with a saltwater solution (1/2 teaspoon of salt in 8 ounces of warm water) after each meal and before bedtime.
3.Soft diet: Patients were recommended to consume a soft diet for one week after surgery to minimize mechanical stress on the implant site.
4.Antibiotics: As a prophylactic measure, patients were prescribed oral amoxicillin (500 mg), to be taken every 12 h for 7 days.
5.Pain and inflammation management: Patients received ibuprofen (400 mg) to manage pain and inflammation, with instructions to take one tablet every 8 h for 4 days.
6.Chlorhexidine rinse: Patients were instructed to rinse their mouth with a 0.12% chlorhexidine gluconate solution twice daily for 2 weeks to enhance plaque control and maintain oral hygiene.
Monitoring of adverse events and patient comfortA standardized protocol monitored intervention-related adverse events. Patients reported discomfort, pain, or unusual symptoms directly to therapists or coordinators. Documentation included event severity, duration, and interventions taken. Therapists monitored patient feedback, adjusting treatment session or providing rest periods as needed to maintain comfort.
Compliance and adherence monitoringTreatment compliance was monitored through comprehensive treatment logs documenting session details and protocol deviations. Patients maintained standardized diaries recording oral hygiene, diet, medication use, and adverse events. Adherence was promoted through appointment reminders and weekly follow-up calls. Each session included standardized questionnaires and oral examinations assessing compliance, hygiene practices, and complications.
Adherence criteria were: LIPUS - attendance of ≥ 75% sessions (6/8) with 20-minute duration; LLLT - completion of all 4 sessions with prescribed parameters; and home care - completion of ≥ 80% prescribed oral hygiene, dietary, and medication protocols (verified through patient diaries). Non-adherence was defined as failure to meet these criteria and analyzed as a binary outcome.
OutcomesPeri-implant marginal bone preservationPeri-implant marginal bone preservation between the bone and implant surface was evaluated using high-resolution digital intraoral radiographs taken with an I-Sensor H2 Digital Intraoral X-ray Imaging System (Guilin Woodpecker Medical Instrument Co., Ltd., Guilin, China) equipped with a rated input of DC 5 V, 500 mA. Radiographs were taken using a paralleling technique with a Rinn film holder and a rigid film-object X-ray source, with settings of 60 kV and 7 mA.
To ensure standardized radiographic imaging across all follow-up visits, a customized positioning device was fabricated for each patient, incorporating a silicone registration material anchored to the existing teeth, with an integrated radiograph holder to maintain consistent beam angulation. The silicone index material was used to create a customized bite block, which helped to maintain the same position of the film and the X-ray tube head for each patient across different time points.
Marginal bone measurements were conducted using specialized imaging software (Vistasoft 2.4.13, Durr Dental Italy S.r.l) [41]. Two key parameters were evaluated:
Radiographic implant length (IL): the linear measurement in millimeters from the coronal implant margin to the apical tip, measured along the implant’s central axis [42].
Residual bone height at the mesial (MI) and distal (DI) aspects of the implant: the vertical distance in millimeters from the coronal implant margin to the first bone-to-implant contact [42].
For each implant, measurements were obtained in triplicate on both mesial and distal aspects, with mean values recorded for each side. A correction factor, calculated from the relationship between actual and radiographic implant dimensions, was applied to compensate for any image distortion [43]. The extent of marginal bone resorption (MBL) was determined by calculating the difference between initial post-surgical bone levels and those observed at the three-month follow-up [44]. Assessment of peri-implant marginal bone preservation was performed at three time points: immediately post-implantation, at the six-week interval, and at three months post-surgery.
Soft tissue healingSoft tissue healing at the implant site was assessed using the Landry Healing Index (HI) immediately post-implant and on days 7, 14, 21, and 30 [45]. This assessment tool examines five key parameters: tissue coloration, granulation tissue presence, bleeding response to palpation, exudate formation, and epithelial integrity. The index utilizes a 1–5 scoring system, where the lowest score [1] represents compromised healing characterized by extensive gingival redness (exceeding 50%), palpation-induced bleeding, evident granulation tissue, failed epithelialization at incision margins with peripheral tissue loss, and presence of suppuration. Conversely, the highest score [5] denotes optimal healing outcomes, indicated by uniformly pink gingival tissue, absence of bleeding upon palpation, no granulation tissue formation, and completely epithelialized incision margins without exposed connective tissue. Superior healing is reflected by higher scores. The scores were recorded by a single calibrated examiner to ensure consistency.
Pain intensityImplant site pain intensity was measured at 12 h post-implant and on days 7, 14, 21, and 30 using the Visual Analogue Scale (VAS), a validated, subjective measure for acute and chronic pain [45]. Patients were instructed to mark their pain intensity on a 10-cm ruler representing a continuum between “no pain” (0) and “worst pain” [10] at the time of assessment [46]. The VAS scores were collected by the patients themselves, and the examiner provided guidance on how to mark the scale correctly.
Oral Health-Related quality of life (OHRQoL)OHRQoL was assessed using the Oral Health Impact Profile-14 (OHIP-14), a shortened version of the OHIP-49 [47]. The OHIP-14 is a widely used instrument to evaluate patients’ satisfaction and OHRQoL after receiving dental implant therapy. The questionnaire was administered in-person by the examiner immediately post-implant, at 6 weeks, and at 3 months [48]. The oral health-related quality of life assessment utilized the OHIP-14 questionnaire, which evaluates seven distinct dimensions: functional restrictions, discomfort/pain, psychological impact, physical limitations, mental well-being, social interactions, and overall life impediment. Each dimension comprises two questions, resulting in a comprehensive 14-item assessment tool. Participants responded using a five-point scale ranging from 0 (never) to 4 (very often). Individual domain scores were obtained by combining the relevant item responses, while the comprehensive OHIP-14 score was calculated as the sum of all domain scores, yielding a potential range of 0–56. An inverse relationship exists between the scores and quality of life, with elevated scores indicating diminished oral health-related quality of life [49].
Follow-up protocolA single calibrated, blinded examiner conducted all assessments following standardized protocols. Baseline measurements (day 0) included marginal bone levels (digital periapical radiographs), soft tissue condition (Landry Healing Index), and OHRQoL (OHIP-14 questionnaire), with pain first assessed via VAS at 12 h post-surgery. Early follow-ups (days 7, 14, 21, 30) evaluated soft tissue healing and pain levels, with all participants (n = 21 per group) completing these assessments without dropouts (Fig. 1).
Comprehensive evaluations at 6 and 12 weeks included marginal bone level radiographs, and OHRQoL assessments. At 6 weeks, one participant from each group missed follow-up (LIPUS: work commitments; LLLT: family emergency; control: lost to follow-up), leaving 20 participants per group. By 12 weeks, additional dropouts occurred in the LIPUS (4 total: relocation, medical issues, loss to follow-up), LLLT (2: perceived lack of benefit, loss to follow-up), and control groups (2: radiograph discomfort, loss to follow-up).
Data analysisANOVA and chi-squared tests were used for baseline comparisons. Data distribution normality was assessed using Shapiro-Wilk test, and homogeneity by Levene’s test. For normally distributed data, MANOVA with repeated measures analyzed time intervals within groups, while MANOVA compared between groups. For non-normal distribution, Friedman and Wilcoxon Signed Ranks tests analyzed within-group differences, while Kruskal-Wallis and Mann-Whitney U tests compared between groups. Significance level was set at p < 0.05 (SPSS v25, IBM).
Comments (0)