
Remember me
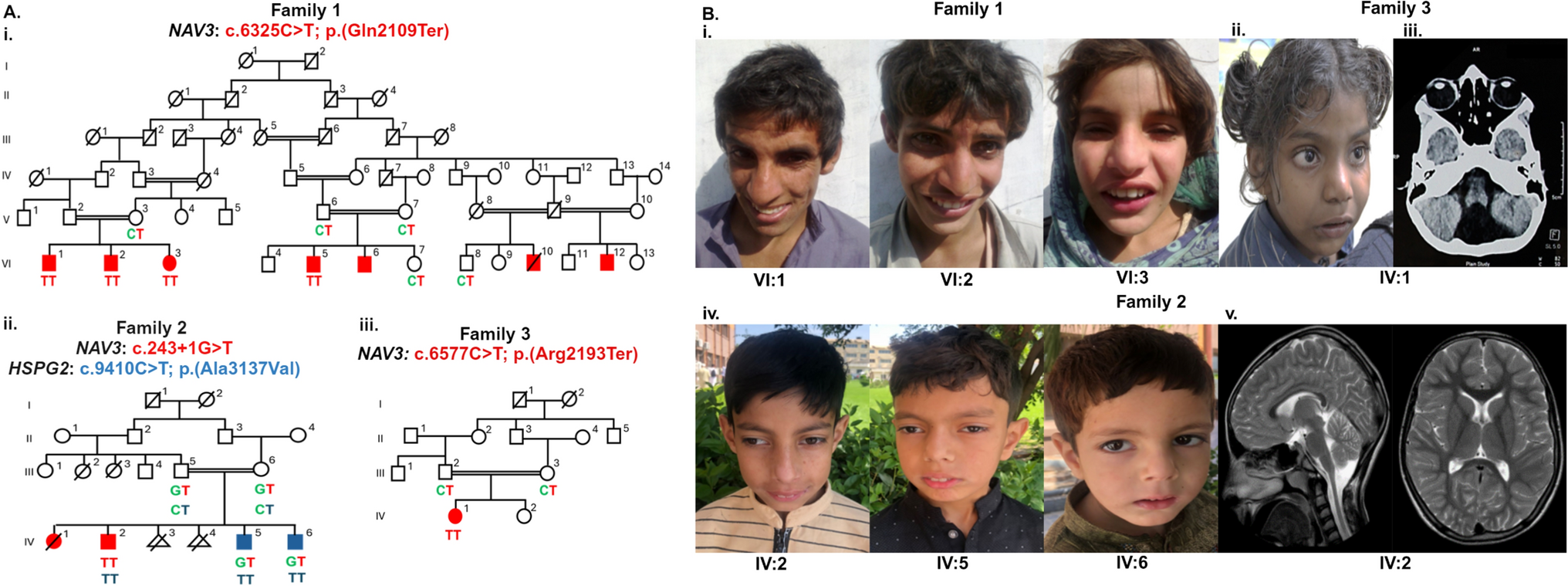



The CAGI6 ID panel challenge was designed to test: (1) the ability of computational methods to predict comorbid phenotypes from targeted gene-panel data, (2) the accuracy of computational methods in predicting causal variants from a set of real genetic data, and (3) the effectiveness of bioinformatics algorithms in improving gene panel data analysis for clinical practice. Additionally, groups may identify variants that were not selected by the Padua NDD Lab, and use them to genetically diagnose the condition of individuals without previous diagnosis.
Summary of the ID panel datasetThe ID panel dataset includes clinical and genetic data from 415 individuals referred to the Padua NDD Lab. Most patients (84.8%) had intellectual disability (ID), and various phenotypic traits were recorded for different portions of the cohort. Autism spectrum disorder (ASD) was present in 49% of the patients and epilepsy in 20%, making these the next most frequent phenotypes. Some patients exhibited multiple phenotypic traits, with 40% showing both ID and ASD. While clinicians provided information about the presence or absence of these phenotypes for most patients, some data were missing (see Fig. 1).
Fig. 1
Summary of CAGI-6 ID panel challenge dataset. a The number of patients where the presence or absence of the phenotype was ascertained by a clinician. b For the 415 patients included in the study, the Padua NDD lab noted at least one variant relevant to the phenotype in 43.4% of the patients
The Padua NDD Lab identified at least one relevant genetic variant in 180 (43,4%) of the 415 sequenced individuals, resulting in a total of 207 variants (see Fig. 1). These variants were categorized based on their potential effects as follows: pathogenic/likely pathogenic (60 variants), variants of uncertain significance (50 variants), and possible risk factors (97 unique variants). Although pathogenic/likely pathogenic and variants of uncertain significance were unique to individuals, some risk factor variants were found in multiple individuals. Combinations of different variant types within the same patient were rare. All variants were treated equally for the purposes of assessing and ranking predictions.
Phenotype prediction assessmentOverall performance metricsThe first part of the ID-challenge in CAGI6 involved predicting the phenotype for each patient among the seven clinical features. The overall submission performance was assessed using the MCC and AUC values (see Fig. 2) and ROC curve (see Fig. 3) for each phenotype. The AUC standard deviation shows the amount of variation expected in each bootstrap iteration. Precision, recall, and F1 score were also computed for each phenotype (see Supplementary Tables S1-S7). Looking at the results presented in Fig. 2, the overall performance of the predictors is quite underwhelming, with AUC values often very close to a random predictor. There is some prediction signal in the ID phenotype, with a maximum AUC value of 0.69 and a standard deviation of 0.04 for SID#2.4. Although improved performance on the ID phenotype might initially be expected due to the bias in the patient panel, the ROC curve and AUC are not directly influenced by the prevalence of the positive class (ID phenotype). Instead, any observed performance improvement would likely result from the model’s ability to distinguish between ID and non-ID cases, rather than the class distribution. Bootstrapping helps estimate the variance in these metrics but does not necessarily increase the AUC value.
Fig. 2
Overall performance for each submission on phenotype prediction. A Each cell represents MCC values. The color scale ranges from green (+ 1, perfect correlation) to red (− 1, negative correlation). White means no better than random prediction. B Each cell represents the mean AUC values of the ROC for 1000 bootstrap iterations. The color scale ranges from dark (+ 1, perfect performance) to white (0, random performance). C Standard deviation (SD) of the bootstrapped AUC values shown in B. AUC, area under ROC curve; MCC, Matthew correlation coefficient; ROC, receiver operating characteristic
Fig. 3
Distribution of the ROC curves for all seven clinical traits. The best performant submission for each phenotype, based on the AUC value, is shown
Performance on different phenotypic traitsThe phenotype prediction assessment was performed individually for each of the seven traits ascertained by clinicians. Figure 4 shows the number of groups that correctly predicted a patient’s phenotype when it was actually present, using again the threshold for maximum FPR of 10%. Of 352 patients with ID in the cohort, 180 (51%) were correctly identified with the ID phenotype by at least three groups, 110 (30%) by two groups, and 53 (15%) by only one group. The ID phenotype appears to be the easiest to predict, with 343 patients (97%) being predicted by at least one group. In comparison, predictions for other phenotypes range from 80% of patients for Ataxia to 94% for Epilepsy. In most cases, only 30% of patients were correctly detected by a single group.
Fig. 4
Performance of the eight groups matching the specific phenotype in 415 patients. Colors represent the proportion and number of groups which correctly predicted the phenotype
For different assigned phenotypic traits (see Fig. 2), the ID phenotype was the easiest to match, with SID#2.4 (AUC 0.69) and SID#5.3 (AUC 0.68) performing well. However, even these top predictors had fairly low MCC values of 0.06 and 0.14, respectively, at a maximum FPR of 10%. The second most prevalent trait in our cohort is ASD, reported by clinicians in 205 out of 415 pediatric patients. The highest AUC values for this phenotype were achieved by SID#1.5 (AUC 0.58), SID#1.2 (AUC 0.56) and SID#5.4 (0.53). It is worth noting that the AUC values for ASD remain close to random.
Comparison of phenotype predictions with CAGI5The overall submission ranking of this challenge was made considering the average AUC rankings for each phenotype (see Table 2). Comparing the results of MCC and AUC to the previous CAGI5 (Carraro et al. 2019) (see Table S10), we do not notice an improvement in phenotype prediction. However, it should be noted that in CAGI5, no bootstrapping of the ROC curves was performed, and the cutoff threshold was calculated by maximizing the MCC values.
Table 2 Ranking of all the predictors based on the ROC AUC values for each phenotypeFor instance, in the case of microcephaly and macrocephaly, where the CAGI6 dataset includes more than twice the number of patients reported with these phenotypes than CAGI5 (see Table 3; Fig. 1), some submissions demonstrated accurate phenotype predictions. SID#6.5 achieved an AUC of 0.64 and a recall of 0.22 for microcephaly (Supplementary Table S5), and SID#1.5 achieved an AUC of 0.61 and a recall of 0.28 for macrocephaly (see Fig. 2 and Supplementary Table S4).
Table 3 Patients for whom Padua NDD Lab identified at least one pathogenic/likely pathogenic, VUS, or Risk factor variant in the answer key, summarized by phenotypeThe CAGI6 cohort reported 71 patients affected by hypotonia, 254 without this phenotype, and 90 for whom information was not available (see Fig. 1). Compared to CAGI5, we did not notice a significant improvement for this phenotype. The maximum AUC across all submissions was 0.55, achieved by SID#1.1 and SID#6.4, attaining a recall of 0.01 and 0.10, respectively (Supplementary Table S6).
The ataxia phenotype was observed in 30 patients, while 285 patients did not exhibit any signs of ataxia (see Fig. 1). The highest-performing model, SID#5.3, has an AUC of 0.63, but fails to attain a maximum FPR of 10% even for the maximum threshold of 1, resulting in a recall and precision equal to 0 (Supplementary Table S7). The AUC results are consistent with the previous assessment. However, it should be mentioned that fewer submissions achieved an AUC score exceeding 0.60 compared to the previous evaluation.
Phenotype prediction in the subset of patients with identified genetic variantsSimilar to CAGI5, we performed an overall phenotype evaluation for patients where the Padua NDD laboratory successfully identified P/LP, VUS, or RF variants. This subset included 180 patients, representing 43.4% of the total cohort (see Fig. 1). The assessment pipeline is the same as before, but now considers only this patient subset. Some changes in prediction performance on the ID phenotype can be appreciated between the entire dataset and this subset, as the percentages of patients predicted by three or more groups raised to 76%, while the overall coverage of predicted patients decreases to 89%. Moreover, we can see an overall improvement regarding the number of groups that correctly identify a patient, with a decrease in the number of patients identified only by one group (see Supplementary Figure S1).
Overall, considering this smaller subset, an improvement of 2.9% was achieved in AUC across the seven phenotypes from all submissions, averaged over all phenotypes. SID#1 achieved the top two positions, while SID#6, which previously held ranks 1 and 3, moved to rank 5 (see Table 2 and Supplementary Table S8).
Variants prediction assessmentThe second part of the CAGI6 challenge was to predict variants associated with the patient phenotype. The overall submission performance was assessed using precision and recall. Figure 5 shows the correctly predicted variants across three classes (P/LP, VUS, RF) for each submission. Groups 8 and 6 correctly predicted most P/LP variants (54 and 52 out of 60, respectively), followed by three other groups (4, 3, 7). Groups 8, 3, and 7 correctly predicted the highest number of VUS and RF variants.
Fig. 5
Predicted variants distribution. Category “Dataset” is the amount of variants which were identified and classified by the Padua NDD lab. Each bar represents the amount of variants and types predicted by each submission. NDD, neurodevelopmental disorder
Figure 6 shows the frequency of each mutation class predicted by different groups. All P/LP variants were predicted by at least two groups (violet), while only 3 were predicted by all groups (green). All VUS were predicted by at least one group, except for the synonymous variant p.Asn839Asn in CNTNAP2, which has not been prioritized by any group. Risk factors are overall very sparsely predicted, with 36% predicted only by one group (29 variants) or not predicted at all (8 variants).
Fig. 6
Performance of the eight groups predicting the correct variants. The amount of variants was calculated for each category (P/LP, VUS, RF). Colors indicate the proportion and number of groups which correctly predicted those variants
Table 4 reports the precision and recall of variant prediction for all submissions. As observed above, SID#8.6 emerges as the most proficient model for capturing a wide range of mutations, exhibiting a recall rate of 82%. However, its precision of 58.7% is lower, implying a significant number of false positives in the results. On the other hand, submission SID#6.2 surpasses all other models with a precision of 72.4%, albeit at a lower recall of 35%, probably due to a limited performance in identifying VUS and RF variants (see Fig. 5). Groups 1, 5, and 2 achieved poor precision and recall in all the three variant classes (see Table 4). This was unexpected, in particular for P/LP variants, considering that the methods developed by group 1 evaluated both the variant frequency and ACMG classification criteria, while group 5 was one of the few to consider filtering variants based on sequencing quality. Additionally, all three groups used the old CAGI5 data to train their methods (see Table 1). Many methods within the same group (e.g. 1, 5, 6) show identical precision and recall as they tend to identify the same variants. This behavior may be due to similarities in the algorithms or criteria used for variant selection.
Table 4 Summary of variants prediction assessment by each submission. Highlighted in bold are the best precision and recall valuesCompared to the CAGI5 challenge, a major improvement can be seen, with the coverage of P/LP variant predictions rising from 64 to 90%, when looking at the respective best model (SID#8.1 for CAGI6 and SID#2.1 for CAGI5). Predictions for VUS and RF variants also improved, rising from 66 to 79% and from 69 to 76%, respectively. Regarding precision for the same models, SID#8.1 achieved 0.516 (Table 4), while in CAGI5 SID#2.1 reached 0.21 (see Table 4 in Carraro et al. 2019).
Challenges in variant predictionMultiple groups indicated some variants to predict the phenotypes which we defined as difficult-to-predict (see Supplementary Table S9). These included variants with sequencing parameters indicating possible technical errors, discordant pathogenicity predictions, and deep intronic variants. Initially, variants were filtered based on sequencing parameters and quality (Aspromonte et al. 2019). Two variants were confirmed as pathogenic after Sanger validation and segregation analysis: p.(Arg504Gln) in GRIN2A, identified as somatic mosaicism by SID#1, 3, 7, and 8, and p.(Pro1585SerfsTer38) in SHANK2, a frameshift deletion initially suspected to be a sequencing error, identified by SID#4 and SID#8.
To prioritize rare missense variants, computational methods were used, including consensus pathogenicity scores from 12 tools and a CADD score (> 25). Although three novel missense variants in PTCHD1, GATAD2B, and ASH1L did not pass this filter, their disease relevance was confirmed through segregation analysis, X-inactivation, and in silico evaluation (Aspromonte et al. 2023). Specifically, for UniPD_0286, the heterozygous GATAD2B variant (c.922T > G; p.Cys308Gly) was identified by six groups. The deep intronic variant MED13L (c.4956–17 A > G) was correctly predicted by four groups. Transcript analysis showed this variant creates a novel cryptic acceptor site, introducing 16 intronic nucleotides into exon 22 (Aspromonte MC et al. 2023).
Re-evaluation and classification of predicted variantsOne of the objectives for the CAGI6 ID panel challenge was to identify variants that might have been missed by the Padua NDD Lab variant analysis but could still be relevant to the patient phenotypes. The Padua NDD Lab reviewed over 8000 variants, including 3016 exonic, 4520 intronic, 7 splicing, and 137 untranslated region (5′/3′-UTR) variants, linked to at least one patient phenotype. Many variants were excluded due to high prevalence in the cohort or general population (gnomAD) or being classified as sequencing errors (see Supplementary Figure S2). Rare variants were reconsidered for Sanger validation, in silico, or functional analysis.
For the female patient UNIPD_0215, Group 1 and 8 indicated the synonymous variant c.240G > A (p.Leu80Leu) in the AP1S2 gene. She was suspected of having Smith-Magenis syndrome, presenting developmental delay, ASD, severe intellectual disability, ataxia, dysmorphisms (e.g., synophrys, large mouth), opposite behavior, and poor impulse control. MRI showed a mega cisterna magna and periventricular ischemic dilatation of the ventricular system. These features align with Pettigrew syndrome (MIM# 304340) caused by mutations in the AP1S2 gene. Human Splicing Finder analysis suggested the variant might alter splicing, leading to its reclassification as likely pathogenic (Aspromonte et al. 2023). However, further segregation and transcript analysis are required to confirm its pathogenicity.
Comments (0)