
Remember me



The global analysis of circRNAs in the FrCx and WB was conducted using previously published datasets of clinical samples from DM1 and DM2 patients, as well as non-DM controls (Otero et al. 2021; Sznajder et al. 2020) (Table S1). Details of the bioinformatics pipeline used for circRNA identification are provided under the Materials and Methods section. Briefly, the RNA samples submitted for sequencing were depleted of rRNA and HMR globin (in blood samples), and neither pre-selection for poly(A)-tailed species nor treatment with RNase R were performed, allowing for an unbiased quantification of both linear and circular RNAs. The transcriptome for each sample was profiled by paired-end, strand-specific capture RNA-seq, with sufficient depth to identify rare circular back-splicing events. To detect the circular BSJ reads from RNA-seq libraries, the CIRI2 pipeline was used (Gao et al. 2018). Custom R scripts were applied to filter the data based on three criteria: “Tier 1 (All)” (circRNAs confirmed by at least two BSJ reads in at least one sample, as defined by the default CIRI2 output), “Tier 2” (circRNAs confirmed by at least five BSJ reads in at least two samples), and “Tier 3” (circRNAs present in all or all but one sample of the analyzed dataset, regardless of the number of BSJs). The two-step filtering process of the CIRI2 output (All) aimed to identify high-confidence BSJs. Ultimately, circRNAs identified in the most stringent filtering category (Tier 3), were considered bona fide transcripts and included in the output files used for further analysis (Tables S2-S5). As shown in Figure S1, regardless of the filtering criteria, the greater number and diversity of circRNAs and their host genes were detected in the DM1 and DM2 samples from the WB, when compared to the FrCx. These genes produced either single or multiple circular transcripts, including their AS isoforms, with multi-circRNA genes (MCGs) being more abundant in the blood than in the cortex. This initial quantitative analysis revealed no positive correlation between the amount of circRNAs and the extent of AAS of linear transcripts, as described in earlier studies (Otero et al. 2021; Sznajder et al. 2020). According to these published data, DM1 and DM2-associated AAS (including CEs, mutually exclusive exons, alternative 5’ and 3’ splice sites, and intron retention) was predominantly detected in the brain of patients. In contrast, the blood from DM2 patients showed less pronounced splicing defects, and no splicing abnormalities were observed in the blood of DM1 patients.
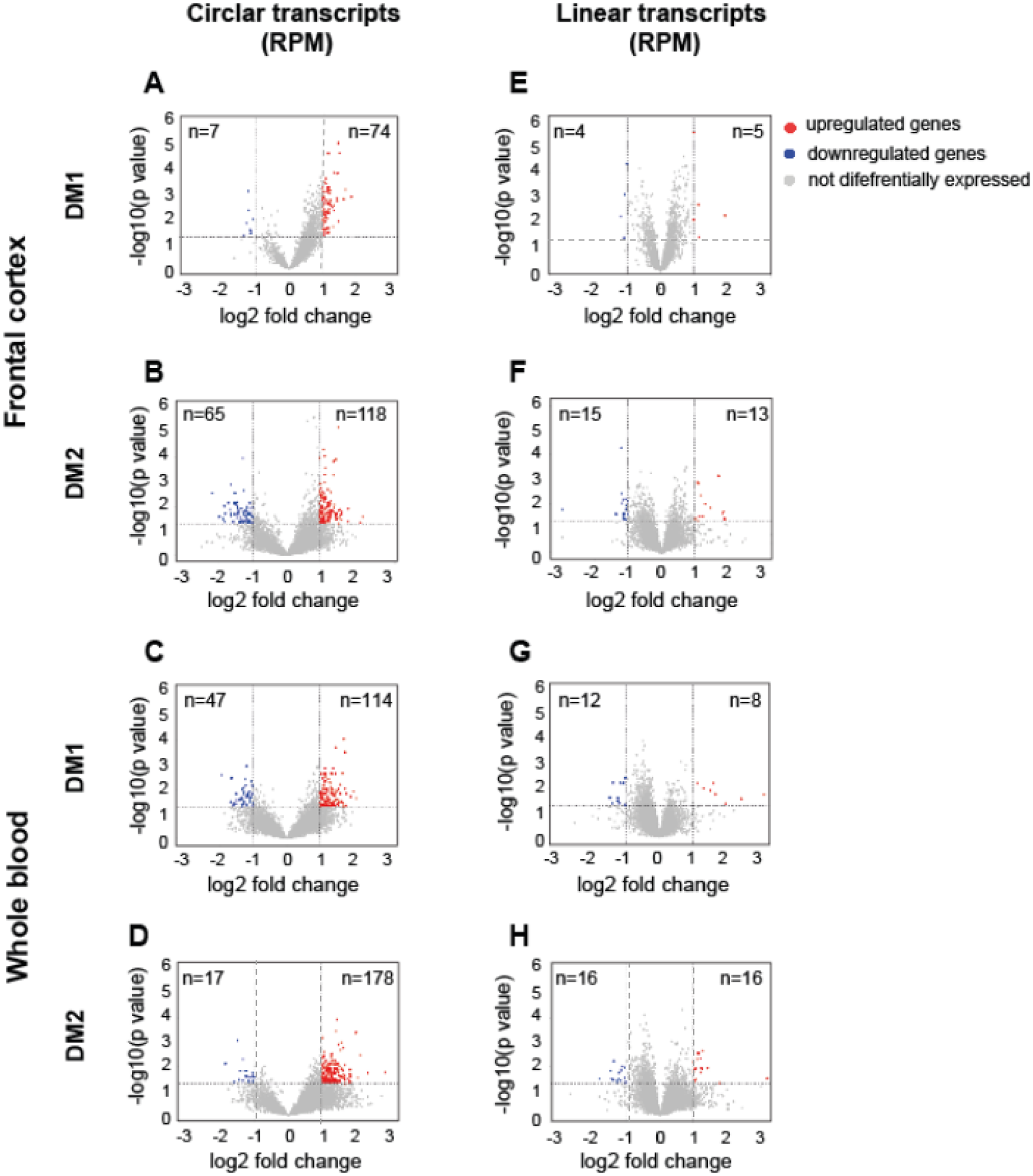
Differential abundance analysis of circRNA expression patterns between diseased and healthy controls was performed in two pairs: DM1 vs. control (sub-set 1) and DM2 vs. control (sub-set 2). The levels of individual circRNAs and their linear cognate transcripts were calculated as the number of BSJ reads and non-junction reads per million mappable reads (RPMs), respectively. This analysis revealed that diseased samples from both tissues exhibited a unique signature of circRNA expression levels, with specific subsets of transcripts being altered in DM1 and DM2 compared to controls. In particular, there was a global upregulation of circRNAs in both neuronal and blood tissues in both types of myotonic dystrophy. These results are detailed in Tables S2-S5 and graphically summarized in Fig. 1. As shown in Fig. 1, among the circular transcripts that exhibited differential expression in DM1 and DM2 (meeting the criteria of log2FC ≤ − 1 or ≥ 1, p < 0.05), 91.4% of circRNAs in the FrCx were upregulated in DM1 samples, and 64.5% in DM2 (chi-squared test, p < 0.0001) (Fig. 1A-B). Similarly, in WB, the upregulated circRNAs remains in the majority and consisted of 70.8% (in DM1) and 91.3% (in DM2) (chi2-squared test, p < 0.0001) (Fig. 1C-D). In all subsets, circRNA abundance was not correlated with the baseline expression of the parental genes (Pearson’s correlation coefficient R² < 0.1) (Fig. 1E-H). While no published data exist on circRNA analysis in DM2, our findings in DM1 are consistent with previously published reports from studies on skeletal muscle samples from DM1 patients and a mouse model of the disease (Czubak et al. 2019a, b; Voellenkle et al. 2019). Both studies highlight the lack of correlation between the levels of circRNAs and their corresponding linear transcripts. This suggests that in different human tissues with a DM1 genetic background, the variability in circRNA abundance cannot be directly explained by the expression levels of their parental genes. Instead, it may be influenced by intrinsic cis-regulatory elements and trans-acting factors, including regulatory splicing mechanisms and varying rates of circRNA stability and turnover in diseased versus control samples. Nevertheless, our study provides further evidence of circRNA dysregulation across various tissues in DM1 and DM2 patients. To validate our RNA-seq findings, we randomly selected a subset of circRNAs for validation in human brain and blood samples. We designed PCR assays with divergent primers for the circRNAs and convergent primers for their linear mRNA counterparts (Figure S2A; Table S6). The size of the circRNA-specific and corresponding linear amplicons was analyzed using agarose gel electrophoresis, and the predicted BSJs were confirmed through Sanger sequencing (Figure S2B-D). All 35 selected circRNAs were successfully validated in these assays.
Fig. 1
The Prevalence of CircRNAs in DM1 and in DM2. Volcano plots depicting differences in the levels of “high-confidence” circRNAs (Tier 3) and their linear host transcripts in DM1 and DM2 samples from FrCx (A, B, E, F) and WB (C, D, G, H). Each red dot (upregulated) and blue dot (downregulated) represent individual transcripts fulfilling the following criteria of expression change: p value < 0.05 and log2 fold change ≤ -1 or ≥ 1 (the thresholds indicated by dotted lines)
Characteristics of differentially expressed CircRNAsThe vast majority of identified circRNAs originated from annotated genes, which generated either single- or multi-circular species as documented in circATLAS (Tables S2-S5). In the FrCx, approximately 8% (in DM1) and 15% (in DM2) of all human protein-coding genes, estimated at 20,000 (Nurk et al. 2022), yielded both circular and corresponding linear transcripts. In contrast, in the WB, these genes comprised about 18% in both DMs. The circRNA genes were found on all chromosomes without any specific preference for their location (Figure S3). The Gene Ontology (GO) analysis of circRNA parental genes (https://david.ncifcrf.gov/home.jsp) revealed distinct functional enrichments. In the FrCx, genes corresponding to circRNAs upregulated in DM1 were enriched in terms related to protein transport, while in DM2, they were enriched in intracellular signal transduction and nervous system development (Figure S4A). In the WB, genes corresponding to DM1-upregulated circRNAs were enriched in transcription and metal ion binding, whereas in DM2, they were enriched in cell cycle and protein binding (p < 0.01) (Figure S4B). Notably, genes corresponding to downregulated circRNAs were not included in the functional association analysis due to their low number.
Based on the CIRI2 output, circRNAs were classified into four major categories: exonic, intronic, intergenic, and mixed (combinations of exonic + intronic, exonic + intergenic, and intronic + intergenic). Both the sense and antisense strands were equally utilized in circRNA generation (Figure S5). Among the differentially expressed circRNAs, the predominant category was exonic circRNAs, which accounted for approximately 90% of Tier 3 circRNAs in the FrCx and 80% in WB. These circRNAs contained either single or multiple exons from a given gene. The second most abundant category was exonic + intronic, which was more enriched in the WB (Figure S5 C-D). Notably, circRNAs of intergenic and mixed intergenic compositions were excluded from further analysis (they comprised less than 1% of the CIRI2 output). Based on circRNA coordinates, their lengths ranged from approximately 60 nucleotides (nt) to over 10,000 nt (Figure S6). The overall distribution of circRNA lengths was similar in both tissues between DM1 and DM2. The most represented length class was 200-1,000 nt (70–76%), with the majority being of exonic composition (99.5% in FC and 68% in WB) (Figure S6 B-E). Considering that the average length of an exon is approximately 200 nt, this suggests that a significant portion of circRNAs represents multi-exonic molecules, while single-exon circRNAs were found to be in the minority (3–6% of Tier 3 circRNAs in the FrCx and WB, respectively). The issue of circularized exon lengths remains a topic of ongoing debate. Some earlier studies have suggested that shorter exons are less likely to circularize, while others argue that smaller exons tend to form circles more readily (Zhang et al. 2014; Szabo et al. 2015). However, our data indicate that as circRNA length increases, the proportion of exonic circRNAs gradually decreases, with circRNAs of mixed composition becoming more prevalent in both DM1 and DM2 across both tissues. This trend was most pronounced in the WB, where mixed (exonic + intronic) circRNAs were most frequently found among transcripts longer than 1,000 nt (Figure S6 D-E). It is important to note that, due to the use of short-read RNA-seq data, our analysis focused on identifying multiple variants of circRNAs based on BSJ coordinates rather than their full lengths. As a result, this approach limits our ability to fully characterize the biogenesis and functional roles of longer circRNAs, particularly those that may involve complex combinations of exons and/or introns due to the AS.
Features of CircRNA-producing lociPrevious studies have suggested that circRNA formation is dependent on both cis-regulatory elements and trans-acting factors, particularly highlighting the sequence requirements in introns flanking BSJs. A critical role in this process has been attributed to intronic Alu repeats derived from short interspersed nuclear elements (SINEs) oriented in opposite directions. Disruption of these repeats has been shown to prevent circRNA formation (Jeck et al. 2013; Liang D and Wilusz JE, 2014; Ivanov et al. 2015). In light of this, we selected the top differentially expressed circRNAs and analyzed the intrinsic genomic features surrounding their BSJs. Using Repeat Masker (https://repeatmasker.org/), we screened for intronic SINEs. Our analysis of DM1 and DM2 samples from both the FrCx and WB revealed that the introns flanking circularized exons were significantly longer than those of non-circularized exons (Fig. 2A-B). These introns contained various Alu elements and exhibited more reverse complementary matches (RCMs) compared to introns from non-circRNA exons (Figure S7; Table S7). Interestingly, these intron features, including the structural stability of the RCMs (measured by the negative free energy value (∆G) for the complementary regions) (data not shown), were shared by both upregulated and downregulated circRNAs. This suggests that while RNA circularization requires similar cis-elements, the regulation of circRNA processing, including their stability, is further modulated by factors that differ between diseased and healthy conditions.
Fig. 2
Features of CircRNA-Producing Loci. (A-B) The length of introns flanking BSJs in differentially expressed circRNAs (log2FC (≤-1.2 & ≥ 1.2) along with size of other introns in the FrCx (A) and WB (B). Red and blue opened and filled circles indicate, respectively, the features of introns in upregulated and downregulated circRNA genes; “up.” and “down.” indicate introns upstream and downstream of circular BSJs, whereas “other” depicts the other introns of circRNA genes away from BSJs; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (two-tail student’s t-test). (C-D) The maximum entropy (MaxEnt) score measuring the strength of splice sites at the 5’ and the 3’ ends of BSJs in individual circRNAs from the FrCx (C) and WB (D). Red and blue circles indicate, respectively, the MaxEnt in individual upregulated and downregulated circRNAs measured at the 5’ ends (filled circles) and 3’ ends (opened circles) of splice sites
It has been suggested that AS is linked to exons that lack canonical splicing signals, and these “weak exons” have been considered a source of genetic susceptibility to splicing errors (de la Mata et al. 2003). To investigate whether this feature might influence splice site (“ss”) selection, leading to exon circularization, we analyzed the strength of the 5’ and 3’ splice sites of BSJs in individual circRNAs, along with the composition of dinucleotides at these junctions. We compared these results with features of other splice sites not involved in circRNA formation. The strength of “ss” was measured using the maximum entropy (MaxEnt) score, developed by Yeo and Burge (Yeo G and Burge CB, 2004). As shown in Fig. 2C-D, the average MaxEnt scores for differentially expressed circRNAs in DM1 and DM2 from the FrCx and WB ranged from 9.1 to 9.8, with no statistically significant differences between upregulated and downregulated circRNAs (t-test). Notably, the MaxEnt scores of the BSJs were not the weakest of the given gene when compared with its other “ss” (Figure S8 A). Additionally, for a few housekeeping genes that do not form circRNAs, the average MaxEnt scores were significantly lower (below 8.0) compared with those of circRNA genes (Figure S8 B). Overall, these results indicate that in DM1 and DM2 from both FrCx and WB, the strength of splice sites at BSJs is unlikely to be a driving factor in circRNA generation.
Cryptic splice sites in exons and introns are involved in CircRNA formation in DM1 and DM2The biogenesis of the vast majority of human exonic circRNAs occurs at the major U2 spliceosome at annotated “ss” flanked by canonical dinucleotides “GU” and “AG” at the 5′ and 3′ ends of an intron, respectively (Zhang et al. 2011, 2022). However, circRNAs processed by the minor U12 spliceosome have also been reported, with BSJs found at unannotated “ss” (Szabo et al. 2015). Our analysis of individual circRNAs from DM1 and DM2 revealed that the majority of BSJs were at canonical donor/acceptor positions, whereas approximately 10–15% (respectively, in the FrCx and in the WB) were located at unannotated “ss” with either canonical or non-canonical dinucleotides (Tables S8 and S9). These BSJs were detected both in exons (ie-BSJs) and in introns (ii-BSJs). The ie-BSJs were often characteristic of the last exons of circRNA genes. Based on the Alternative Splice Site Predictor (ASSP) tool (http://wangcomputing.com/assp/) (Wang M and Marin A, 2006), they were categorized as cryptic, constitutive, or unclassified donors or acceptors. Thus, splice sites in exons are involved in back-splicing, leading to the production of circRNAs that contain partial exon sequences. Interestingly, we identified that cryptic “ss” contributing to circRNA biogenesis are located either in the last coding exons (e.g., hsa_CPNE1_0001) or in the 3′-UTR (e.g., hsa_ZNF250_0004) (Figure S9; Table S8). Based on previously published human transcriptomics data, the non-canonical splicing signals inside annotated protein-coding exons were attributed to cryptic introns called exitrons (Marquez et al. 2015). Exitrons are unique in that they have both protein-coding (exon) and splicing (intron) potentials while also possessing canonical splice-site signals. However, the first exons of circRNA genes have also been identified with non-canonical “ss,” and these were classified as cryptic acceptors (Table S8). Thus, terminal exons, which lack their 3′ or 5′ splicing signals and would normally prevent them from circularizing, overcome this obstacle and become donors or acceptors of new cycles of the AS.
The second type of unannotated “ss” was found in introns and was typical either of purely intronic (e.g., hsa-ROBO2_0044) or exonic-intronic circRNAs (e.g., hsa-RASGRF2_0029) (Table S9). The latter group was the majority, regardless of the subsets analyzed, and this category of circRNAs predominates in the blood compared with the cortex. Over 61% of intronic circRNAs have their “ss” in the first or second introns, whose lengths range from a few thousand to over 1 million nucleotides. About 30% of these circRNAs have cryptic “ss” marked by non-canonical dinucleotides, with MaxEnt scores ranging from 2.45 to 14.43 (Table S9). Further characteristics of circRNAs with canonical and non-canonical “ss” involved in the formation of BSJs revealed significant differences in their levels, including those of their host linear transcripts. Overall, circRNAs originating from the utilization of unannotated splice sites were less abundant compared to those with canonical donor/acceptor “ss” (Figure S11). This observation suggests that the biogenesis of different types of circRNAs is influenced by divergent pathways and transcript isoforms, with ongoing experiments aimed at validating this hypothesis (Mroczko-Młotek A, manuscript in preparation).
Altogether, these results indicate that the utilization of cryptic splice sites in specific segments of circRNA genes contributes to their biogenesis in different tissues from DM1 and DM2. Cryptic “ss” in introns lead to the enrichment of the circRNA population containing partial intronic sequences, which are predominantly identified in blood. This suggests that circRNA formation involves tissue-specific splicing mechanisms that use both canonical and cryptic splice sites, warranting further investigation.
Circularized exons are not among cassette exons aberrantly spliced in DM1 and DM2More than 95% of genes in the human genome are alternatively spliced, and one of the most common types of AS events is CE, also known as exon skipping (Dvinge H et al., 2015). In vitro and in vivo studies in flies and vertebrates have identified several characteristic features of CEs that differentiate them from constitutive exons. Specifically, CEs are shorter than constitutively spliced exons, have longer flanking introns, lower GC content, and weaker splice sites (Sorek et al. 2004; Rukov et al. 2007; Bell et al. 1998; Fox-Walsh et al. 2005; Kim et al. 2007; Gelfman et al. 2012). These associations suggest that genome architecture contributes to variations in AS types and frequencies. However, the enrichment of trans-acting factor binding in introns flanking the alternative exons also influences their splicing fate, as demonstrated in DM1 and DM2 studies (Savkur et al. 2001; Wang et al. 2019). Thus, we aimed to determine whether the above features also characterize exons that escape forward splicing by forming single-exon circRNAs (SE-circRNAs). SE-circRNAs comprised about 3–5% of the highly confident circRNAs identified in the FrCx and WB. We compared this result with the aberrant splicing of CEs described previously in these two tissues (Otero et al. 2021; Sznajder et al. 2020). First, we found no common exons between SE-circRNAs and CEs that were aberrantly spliced in DM1 and DM2 for a given gene. Second, our results showed a consistent relationship between gene architecture and the occurrence of either SE-circRNAs or CEs; however, these events did not share similar characteristics (Fig. 3). In particular: (i) exons of SE-circRNAs were always significantly longer than CEs; (ii) SE-circRNAs were flanked by much longer introns, mostly in the FrCx; and (iii) the strength of the 5′ and 3′ “ss,” as measured by MaxEnt scores, was significantly higher in SE-circRNAs from the FrCx, whereas in WB, there was no such diversity. Thus, in the FrCx from DM1 and DM2, heterogeneity in the splice sites influenced the selection of an AS type, with exons having more poorly defined intron-exon boundaries being more susceptible to CE events, whereas those with stronger “ss” were more likely to undergo circularization.
Fig. 3
Features of Single-Exon CircRNAs and Misspliced Cassette Exons. The comparison of exon size (A), its flanking introns lengths (B), and splice sites strength (MaxEnt) (C) between single-exon circRNAs and CEs aberrantly spliced in the FrCx and WB (based on published results from Otero et al. 2021; and Sznajder LJ et al., 2020). *p < 0.05, **p < 0.005, ***p < 0.0005 (two-tail student’s t-test)
In searching for factors associated with exon circularization, we also analyzed the binding signature of splicing factors in regions flanking BSJs of SE-circRNAs and compared them with those of CEs aberrantly spliced in DM1 and DM2 (Otero et al. 2021; Sznajder et al. 2020). Based on published data obtained from CLIP analysis of MBNL1 and CUGBP1 proteins, their binding to upstream or downstream regions of alternative exons stimulates antagonistic effects (Wang et al. 2012). While MBNL1 promotes alternative exon exclusion when binding to the cassette and upstream of it, CUGBP1 achieves the same effect when binding downstream (Masuda et al. 2012). This scenario is disrupted in myotonic dystrophies due to diminished functional levels of MBNL1 and upregulation of hyperphosphorylated CUGBP1, which is predominantly found in DM1 (Kuyumcu-Martinez et al. 2007; Wang GS and Cooper TA, 2007). Using the SpliceAid database of human splicing factors and their RNA-binding sites (Giulietti et al. 2013), we searched for binding enrichment of these proteins in three distinct regions: the circularized exon itself, and 250 nucleotides downstream and upstream in the flanking introns. For MBNL1, we identified 5-mer motifs (YGCY), including UGCUU and GCUGC, while for CUGBP1, the motif was UG-rich (Wang et al. 2012; Lambert et al. 2014). Among the differentially expressed SE-circRNAs from the FrCx and WB, we did not find exons with a strong signature for MBNL1 binding characteristic of exon exclusion. However, in the cortex from DM1, we identified enrichment of CUGBP1 binding in the introns flanking BSJs of some upregulated SE-circRNAs (e.g., hsa-RC3H1_0001 and hsa-DCLK2_0001 had 96 and 62 CUGBP1 binding sites, respectively, within 250 bp downstream of the circularized exons). Interestingly, elevated levels of hsa-RC3H1_0001 were also detected in DM1 skeletal muscle, suggesting that upregulation of CUGBP1 in these two DM1 tissues may affect the biogenesis of some circRNAs. In support of this assumption, no statistically significant differences in the levels of these two circRNAs were found in the WB or in any tissues from DM2. The potential involvement of CUGBP1 in RNA circularization is currently under investigation (Srinivasan A, manuscript in preparation).
Altogether, these results reveal distinct features of circularized exons that differentiate them from constitutive and alternatively spliced exons in DM1 and DM2. The involvement of MBNL1 in the biogenesis of circRNAs, which was questioned in a previous study of DM1 skeletal muscles, is supported by the current analysis. However, our preliminary results suggest that the circularization of some RNAs could be influenced by CUGBP1 protein, which is primarily implicated in DM1 pathogenesis.
Comments (0)