Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nature Methods 7(4):248–249
Article
CAS
PubMed
PubMed Central
Google Scholar
Alirezaie N, Kernohan KD, Hartley T, Majewski J, Hocking TD (2018) ClinPred: prediction tool to identify disease-relevant nonsynonymous single-nucleotide variants. The American Journal of Human Genetics 103(4):474–483
Article
CAS
PubMed
Google Scholar
Ancien F, Pucci F, Godfroid M, Rooman M (2018) Prediction and interpretation of deleterious coding variants in terms of protein structural stability. Scientific Reports 8(1):4480
Article
PubMed
PubMed Central
Google Scholar
Bergquist T, Stenton SL, Nadeau EA, Byrne AB, Greenblatt MS, Harrison SM, Tavtigian SV, O’Donnell-Luria A, Biesecker LG, Radivojac P, et al. (2025) Calibration of additional computational tools expands ClinGen recommendation options for variant classification with PP3/BP4 criteria. Genetics in Medicine
Brandes N, Goldman G, Wang CH, Ye CJ, Ntranos V (2023) Genome-wide prediction of disease variant effects with a deep protein language model. Nature Genetics 55(9):1512–1522
Article
CAS
PubMed
PubMed Central
Google Scholar
Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R (2009) Functional annotations improve the predictive score of human disease-related mutations in proteins. Human Mutation 30(8):1237–1244
Article
CAS
PubMed
Google Scholar
Carter H, Douville C, Stenson PD, Cooper DN, Karchin R (2013) Identifying mendelian disease genes with the variant effect scoring tool. BMC Genomics 14(3):1–16
Google Scholar
Cheng J, Novati G, Pan J, Bycroft C, Žemgulytė A, Applebaum T, Pritzel A, Wong LH, Zielinski M, Sargeant T et al (2023) Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381(6664):7492
Article
Google Scholar
Chennen K, Weber T, Lornage X, Kress A, Böhm J, Thompson J, Laporte J, Poch O (2020) MISTIC: A prediction tool to reveal disease-relevant deleterious missense variants. PLoS One 15(7):0236962
Article
Google Scholar
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the functional effect of amino acid substitutions and indels. PLoS One 7(10):46688
Article
Google Scholar
Chun S, Fay JC (2009) Identification of deleterious mutations within three human genomes. Genome Research 19(9):1553–1561
Article
CAS
PubMed
PubMed Central
Google Scholar
Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, Lu X (2012) Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Frontiers in Genetics 3:35
Article
PubMed
PubMed Central
Google Scholar
Cingolani P, Platts A, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6(2):80–92
Article
CAS
PubMed
PubMed Central
Google Scholar
Critical Assessment of Genome Interpretation Consortium (2024) CAGI, the Critical Assessment of Genome Interpretation, establishes progress and prospects for computational genetic variant interpretation methods. Genome Biology 25(1):53
Article
Google Scholar
Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S (2010) Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Computational Biology 6(12):1001025
Article
Google Scholar
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X (2014) Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Human Molecular Genetics 24(8):2125–2137
Article
PubMed
PubMed Central
Google Scholar
Feng B-J (2017) PERCH: a unified framework for disease gene prioritization. Human Mutation 38(3):243–251
Article
CAS
PubMed
PubMed Central
Google Scholar
Frazer J, Notin P, Dias M, Gomez A, Min JK, Brock K, Gal Y, Marks DS (2021) Disease variant prediction with deep generative models of evolutionary data. Nature 599(7883):91–95
Article
CAS
PubMed
Google Scholar
Freedman D, Diaconis P (1981) On the histogram as a density estimator: \(_\) theory. Zeitschrift für Wahrscheinlichkeitstheorie und verwandte Gebiete 57(4):453–476
Article
Google Scholar
Gao H, Hamp T, Ede J, Schraiber JG, McRae J, Singer-Berk M, Yang Y, Dietrich AS, Fiziev PP, Kuderna LF et al (2023) The landscape of tolerated genetic variation in humans and primates. Science 380(6648):8153
Article
Google Scholar
Garber M, Guttman M, Clamp M, Zody MC, Friedman N, Xie X (2009) Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 25(12):54–62
Article
Google Scholar
Ghosh R, Harrison SM, Rehm HL, Plon SE, Biesecker LG (2018) ClinGen Sequence Variant Interpretation Working Group: Updated recommendation for the benign stand-alone ACMG/AMP criterion. Human Mutation 39(11):1525–1530
Article
PubMed
PubMed Central
Google Scholar
Grimm DG, Azencott C-A, Aicheler F, Gieraths U, MacArthur DG, Samocha KE, Cooper DN, Stenson PD, Daly MJ, Smoller JW, Duncan LE, Borgwardt KM (2015) The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Human Mutation 36(5):513–523
Article
PubMed
PubMed Central
Google Scholar
Hu Z, Yu C, Furutsuki M, Andreoletti G, Ly M, Hoskins R, Adhikari AN, Brenner SE (2019) VIPdb, a genetic Variant Impact Predictor Database. Human Mutation 40(9):1202–1214
Article
PubMed
PubMed Central
Google Scholar
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D et al (2016) REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. The American Journal of Human Genetics 99(4):877–885
Article
CAS
PubMed
Google Scholar
Ionita-Laza I, McCallum K, Xu B, Buxbaum JD (2016) A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nature Genetics 48(2):214–220
Article
CAS
PubMed
PubMed Central
Google Scholar
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, Bernstein JA, Bejerano G (2016) M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nature Genetics 48(12):1581–1586
Article
CAS
PubMed
Google Scholar
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581(7809):434–443
Article
CAS
PubMed
PubMed Central
Google Scholar
Katsonis P, Wilhelm K, Williams A, Lichtarge O (2022) Genome interpretation using in silico predictors of variant impact. Human Genetics 141(10):1549–1577
Article
PubMed
PubMed Central
Google Scholar
Katz AE, Nussbaum RL, Solomon BD, Rehm HL, Williams MS, Biesecker LG (2020) Management of secondary genomic findings. The American Journal of Human Genetics 107(1):3–14
Article
CAS
PubMed
Google Scholar
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols 4:1073–1081
Article
CAS
PubMed
Google Scholar
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W et al (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Research 46(D1):1062–1067
Article
Google Scholar
Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, Mooney SD, Radivojac P (2009) Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25(21):2744–2750
Article
CAS
PubMed
PubMed Central

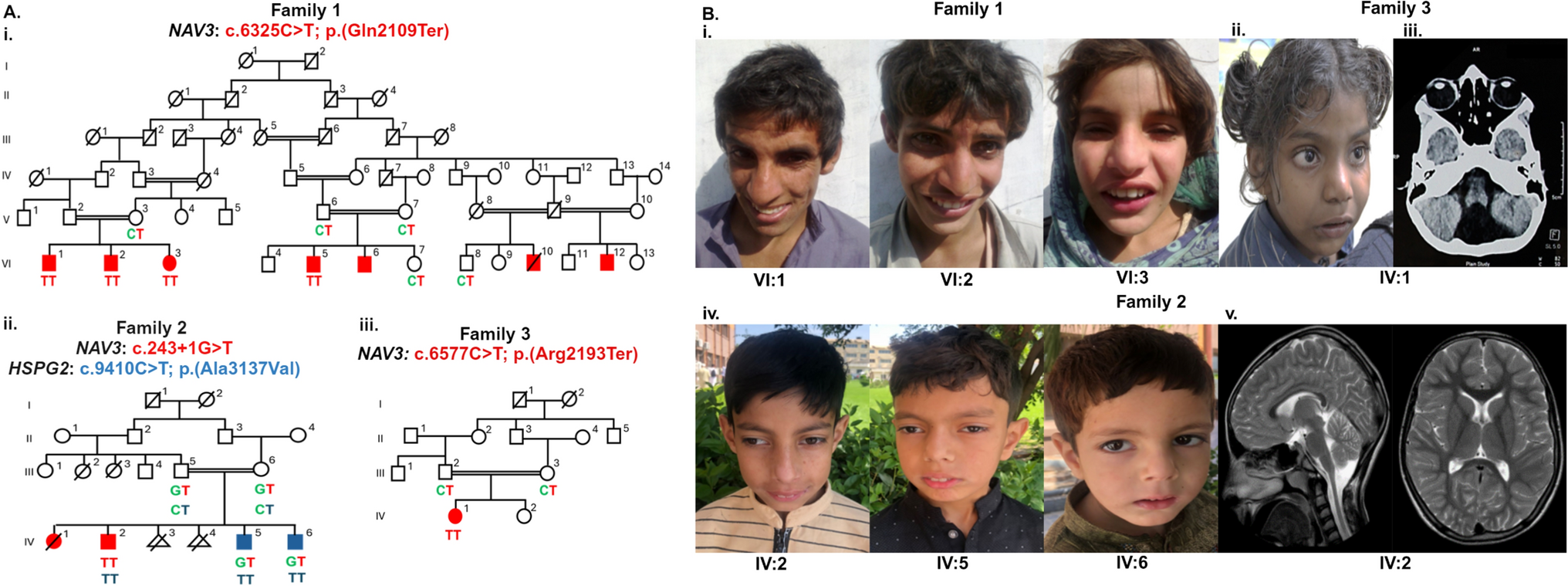

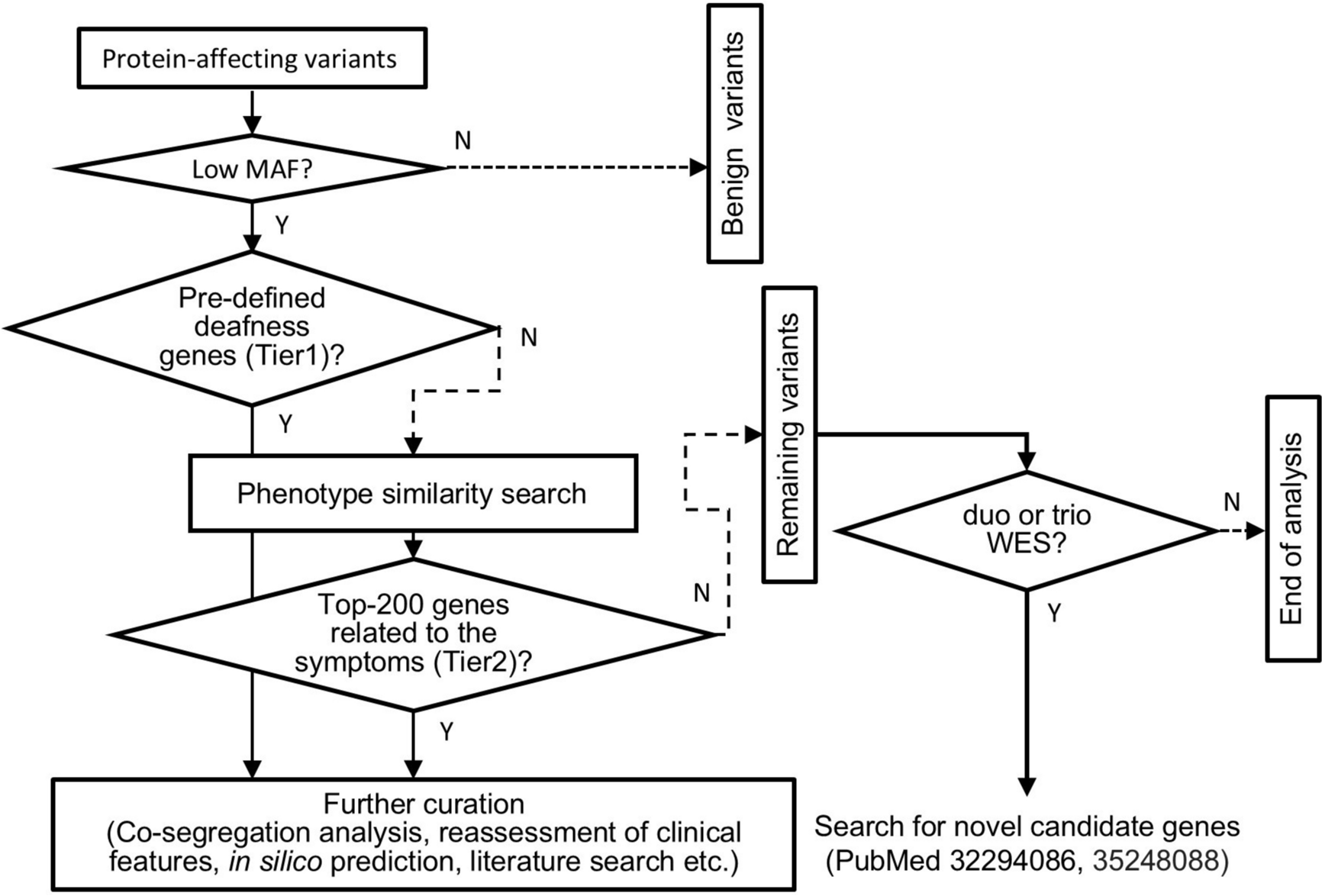
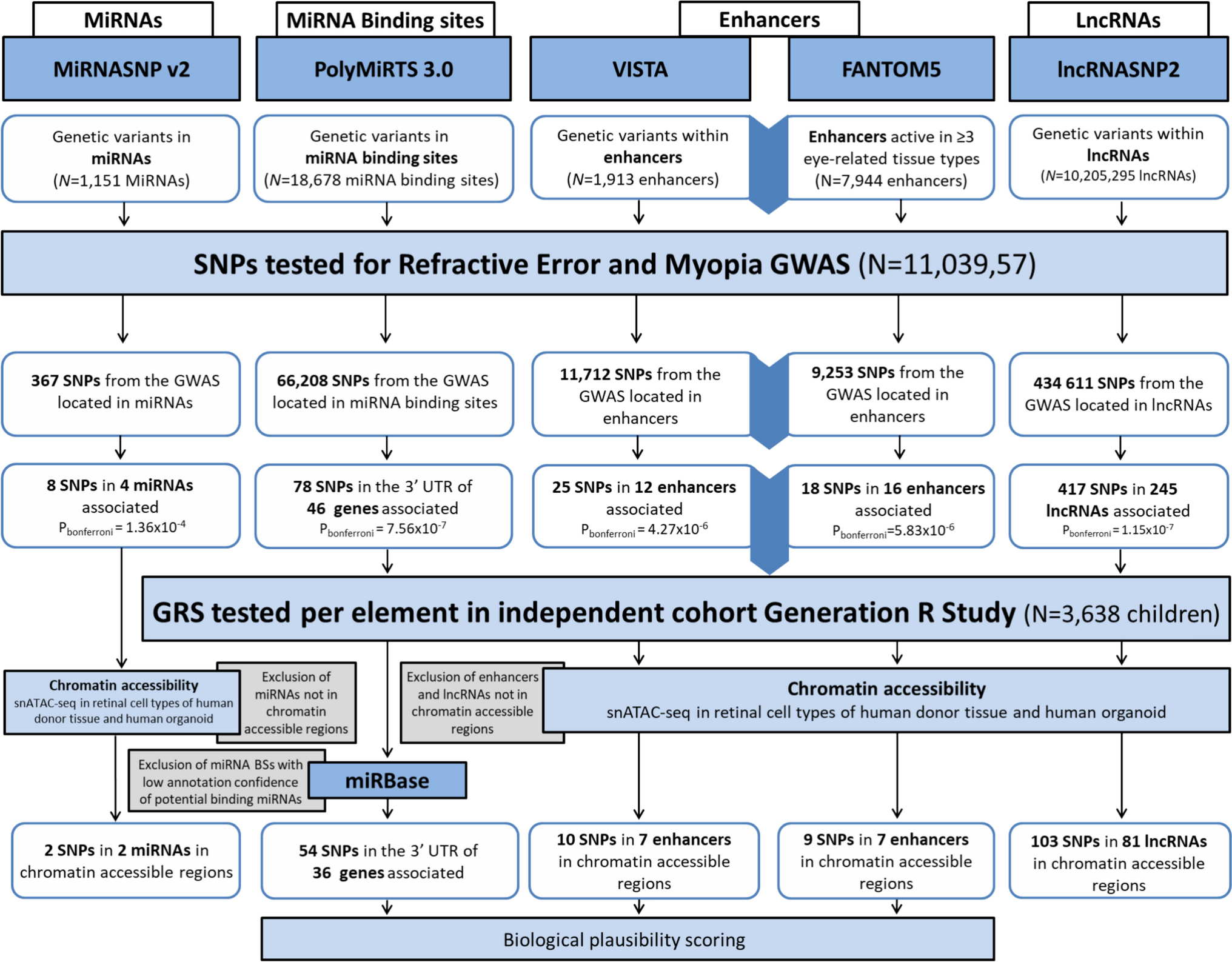

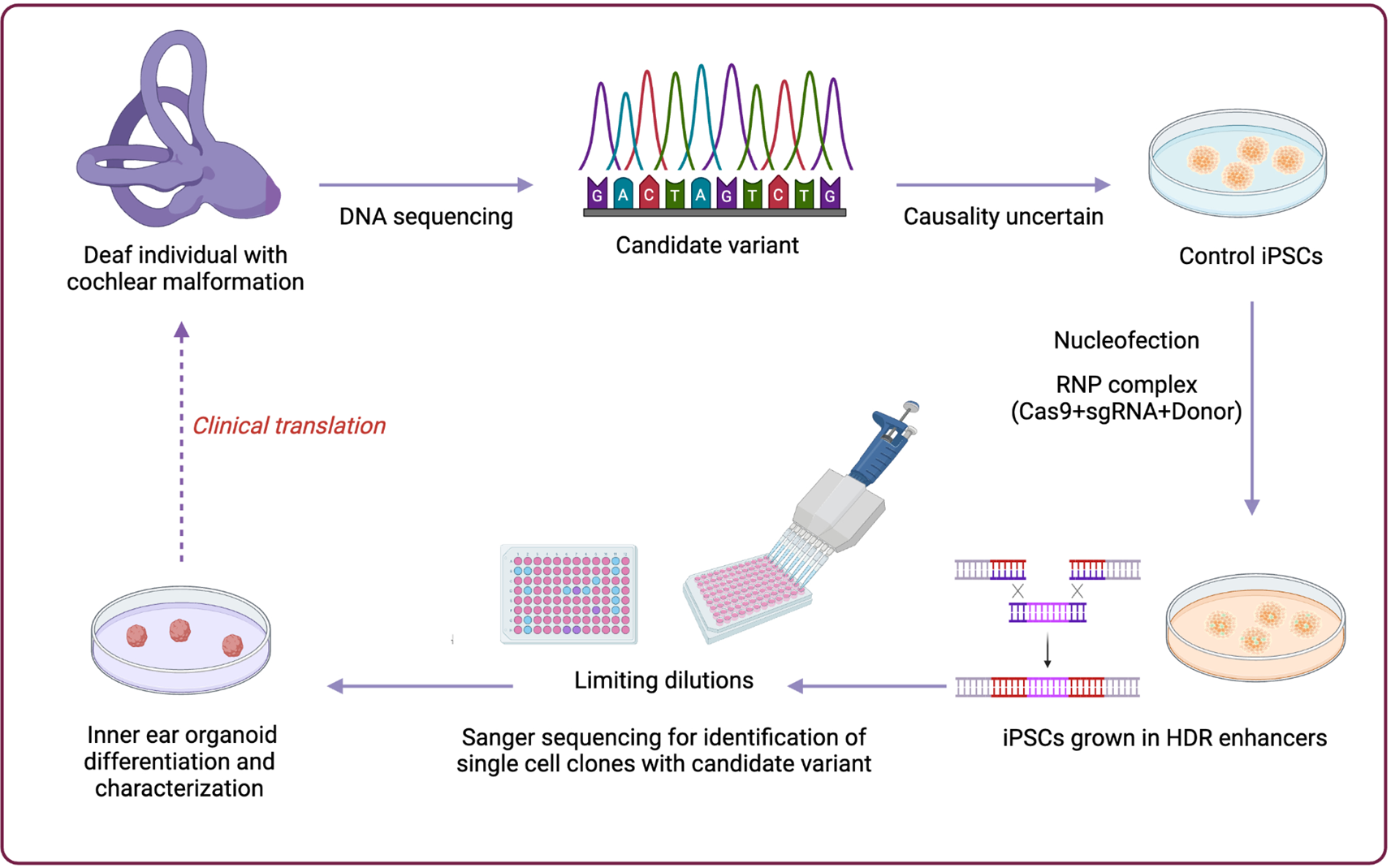

Comments (0)