
Remember me
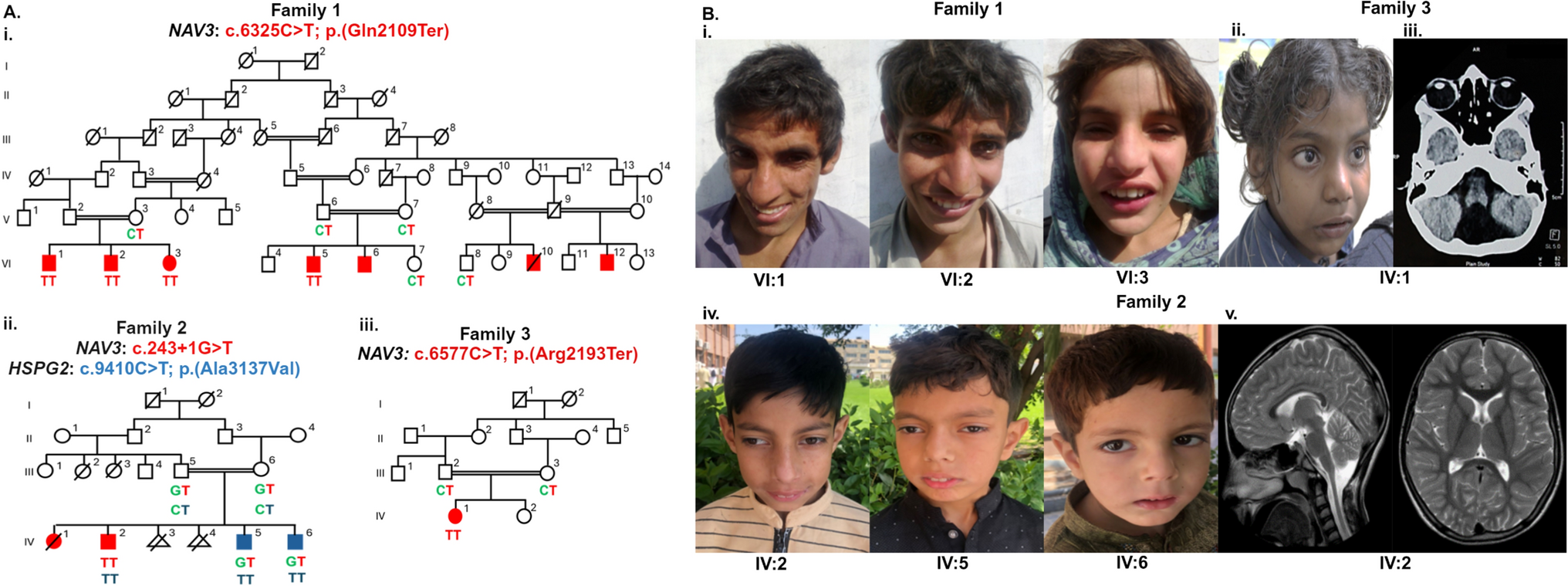

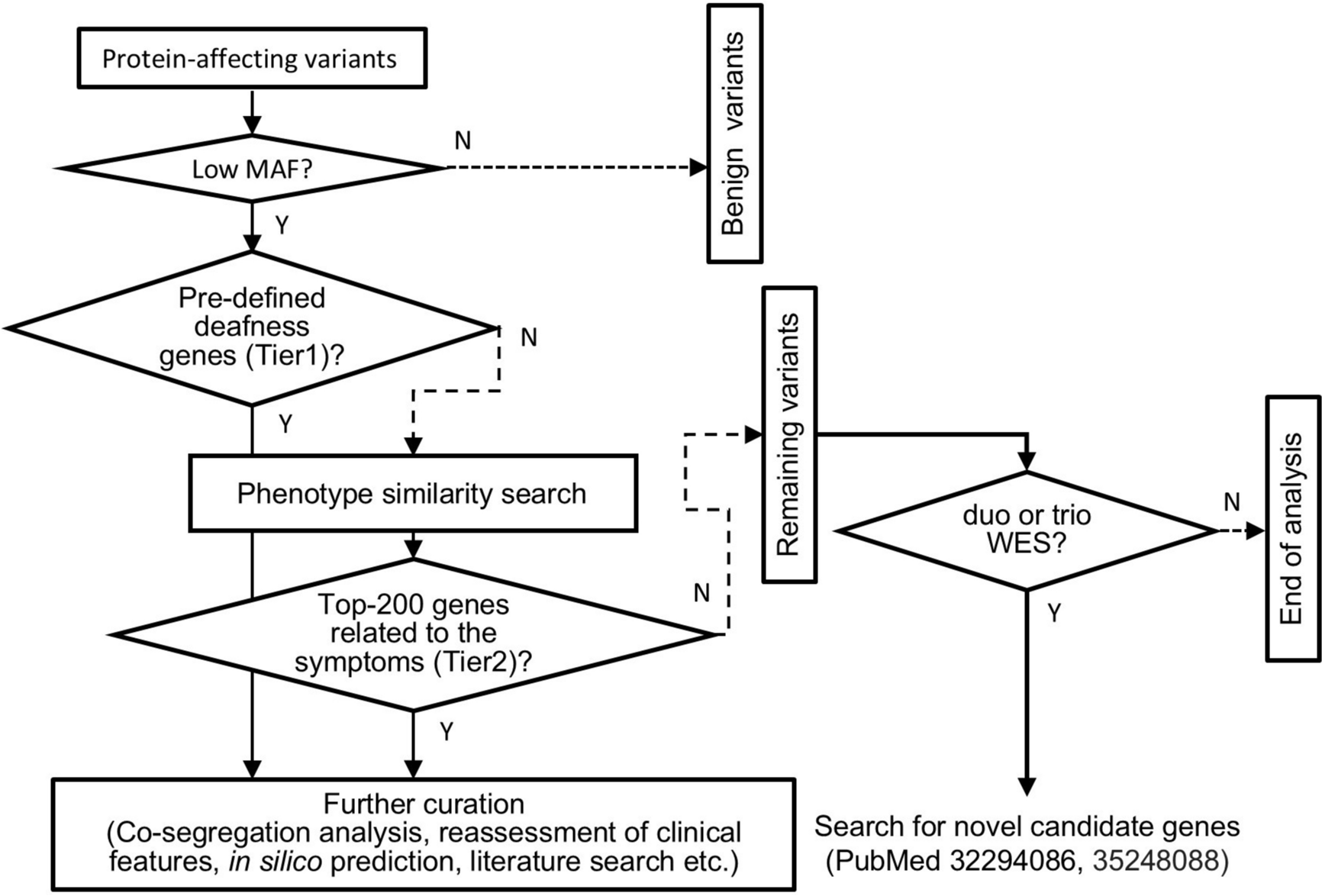
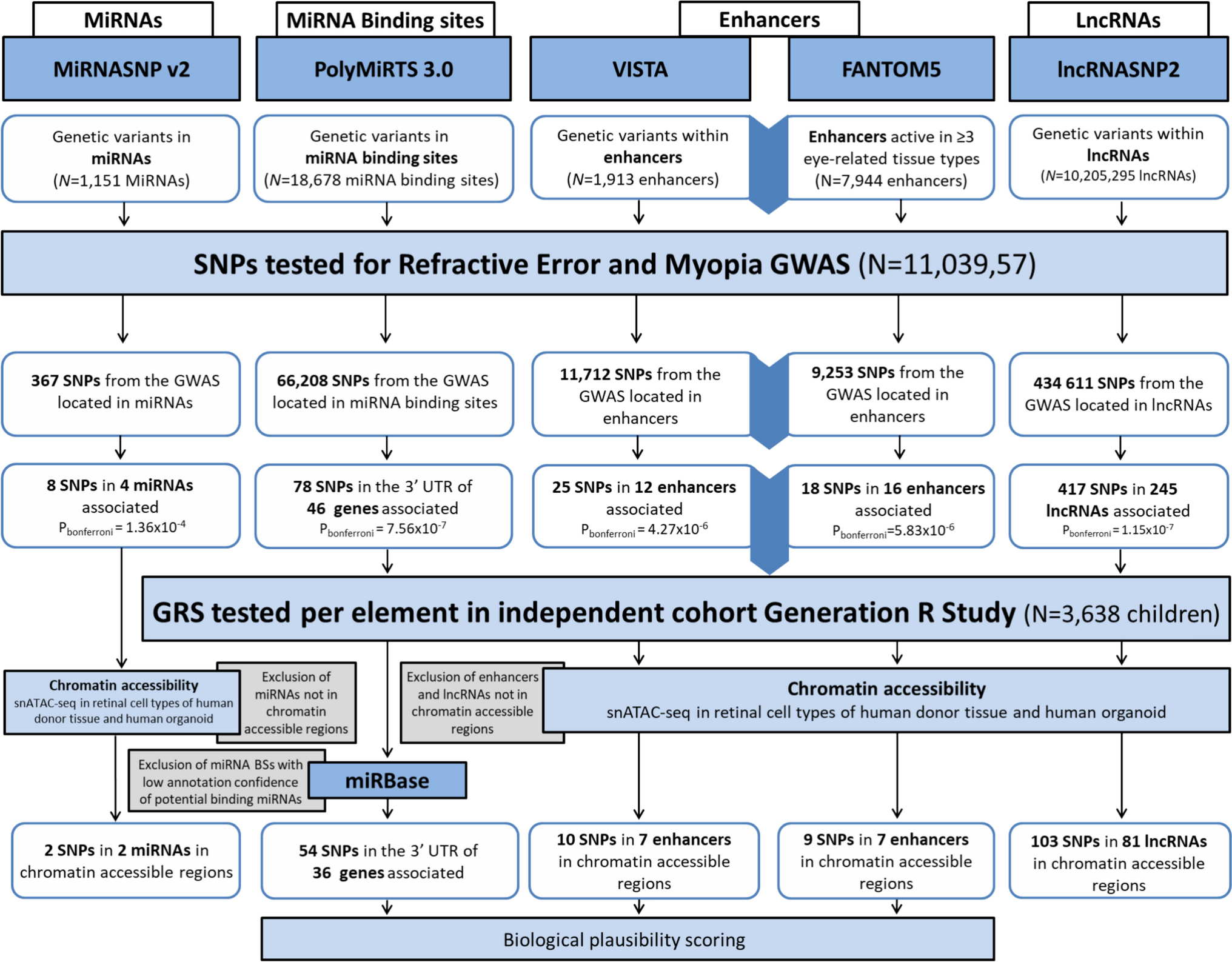

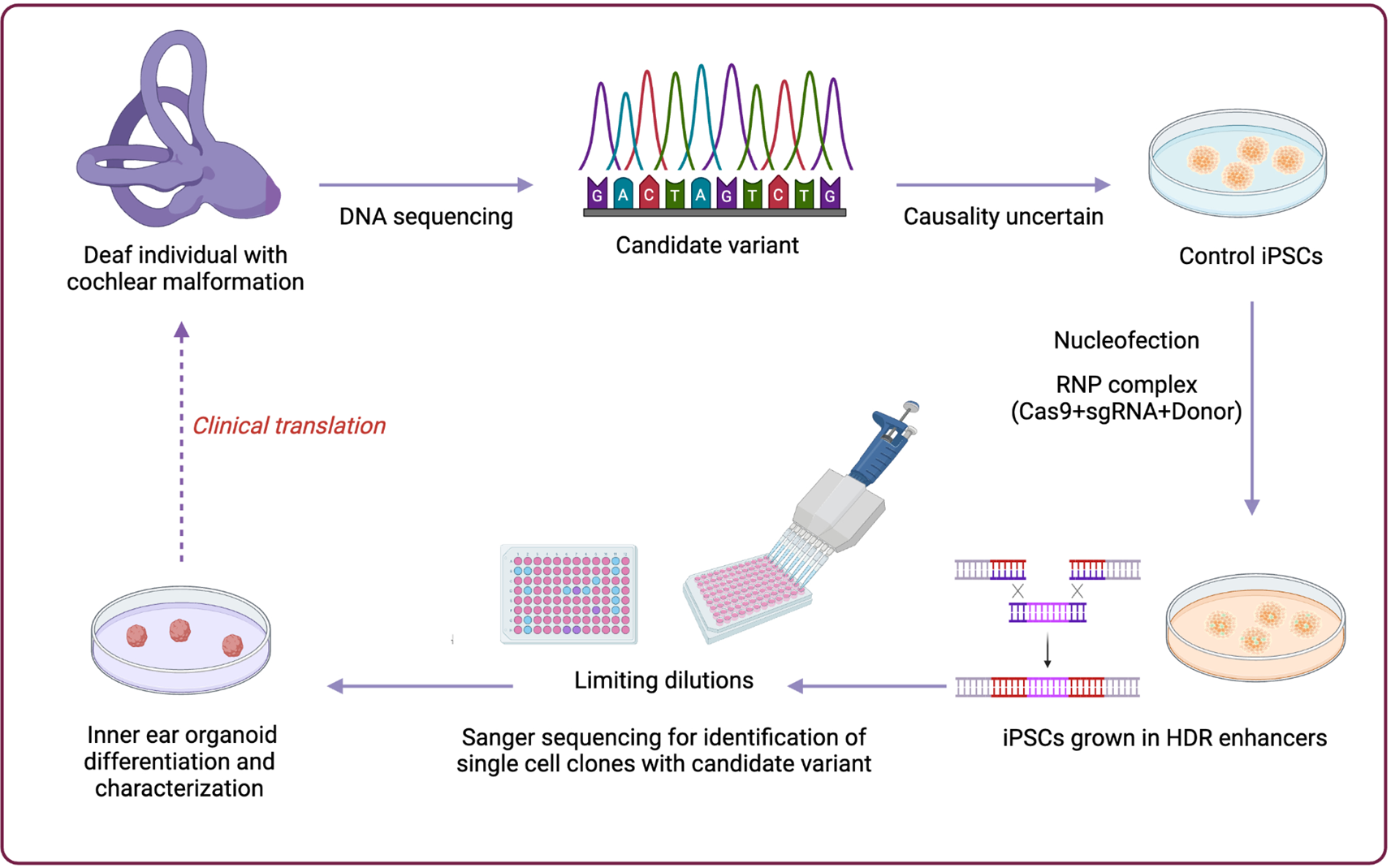

Probands with a clinical tentative diagnosis of RP together with their affected and unaffected relatives were referred from Grantham Hospital under the Hospital Authority of Hong Kong SAR, to the HKGP, with written informed consent obtained. We primarily collected whole blood samples, with buccal mucosa or saliva samples used as alternatives when whole blood collection was not feasible for the participants.
Short read GS and data processing and filteringSamples were prepared and sequenced by Illumina NovaSeq 6000 sequencer, followed by GATK best practice for data processing. Data was further prepared according to Fig. 1A. Selected genes were listed in Supplementary Table S1. Please refer to the Supplementary file for detailed description.
Fig. 1
Data processing workflow comparison flowchart of HKGP. A Data preparation. Filtered variant list was prepared from sequencing GS reads accordingly. B Details of the three steps in S-BCAW. C Details of the two steps in MICAW. Curation conclusions and individual ACMG criteria assigned were compared between two workflows before reporting
Manual individual case analysis workflow (MICAW)All variants in the filtered list from a single case with related family members were curated and classified by experienced reviewers in two rounds according to the standard ACMG/AMP guidelines, SVI Working Group Recommendations, along with ClinGen Variant Curation Expert Panels (VCEP) when applicable (Richards et al. 2015) (Fig. 1C). Different trained and experienced genome curators, who adopted the role as “Reviewer 1”and “Reviewer 2” respectively were responsible for the tertiary analysis process.
The first round of manual variant curation by “Reviewer 1” started with assignment of variant specific ACMG criteria, namely PM2, PP2, and PP3. PM2_supporting was only assigned to variants that are rare (gnomAD East Asian allele frequency < = 0.005) according to the SVI Recommendations (Chen et al. 2024). PP2 was assigned to missense variants where the gene missense constraint z-score from gnomAD database was above 3.09 and the regional missense constraint was significant (p < 1e-3) (Chen et al. 2024). PP3 was assigned according to the computational REVEL score according to the ClinGen Recommendations for PP3/BP4 criteria (Ioannidis et al. 2016; Pejaver et al. 2022). Publication or case–control based criteria such as PS4, PS1 and PM3, were gathered from various sources including ClinVar reported cases and literature searches. PVS1 was determined by manually checking the relevant factors according to the decision tree in ClinGen PVS1 recommendation (Tayoun et al. 2018). PM4 was assigned to variants that affected protein length without altering the reading frame. Family-based criteria, PS2 and PP1, were carefully assigned by manually verifying and confirming the variant status of the parents. All criteria were applied with respect to strength modifications as recommended by ClinGen SVI working groups (Rivera‐Muñoz et al. 2018).
A detailed second-round curation by “Reviewer 2” validated and confirmed the assigned ACMG criteria of candidate variants. This meticulous review process, conducted in collaboration by two reviewers, ensured precision and reliability. Additionally, this approach has proven effective in diagnosing previously undiagnosed diseases in our institute. Selected candidate variants, including compound heterozygous and homozygous pathogenic or likely pathogenic (PLP) variants in recessive genes, and single PLP variants in dominant genes, were then validated through orthogonal sequencing to confirm the diagnostic yield of the cohort.
Semi-automated bespoke cohort analysis workflow (S-BCAW)All filtered variants from the same cohort were merged into a single variant list, with sample ID as additional label. Selected criteria from ACMG/AMP guidelines for sequencing variant interpretation were calculated automatically, together with variant information collection for additional criteria. Variants were then classified into different actionable categories, and further reviewed by experienced reviewers to verify the details of the assigned criteria and classifications (Fig. 1B).
S-BCAW Step 1: Automatic assignation of selected ACMG criteriaThe following ACMG criteria were automatically assigned based on collected resources, or preassigned using partially collected data for each variant, to facilitate further processing.
PVS1: For predicted loss-of-function variants, the AutoPVS1 tool, developed in accordance with the ClinGen Sequence Variant Interpretation (SVI) Working Group guidelines for the interpretation of the loss-of-function (LoF) PVS1 ACMG/AMP variant criterion, has been incorporated into our analytical workflow (Xiang et al. 2020). This integration enables the systematic evaluation of the PVS1 criterion level across various genomic alterations, encompassing start/stop gain/loss SNVs, small frameshift indels, splicing variants, and multi-exon SVs. Furthermore, our workflow meticulously extracts detailed information based on PVS1 decision tree, thereby elucidating the foundational data supporting the determination of PVS1 criterion levels.
PM2: To evaluate the rarity of variants in the population, disease-specific PM2 thresholds for population allele frequency, pertinent to both dominant and recessive genes within the cohort, were initially established through a manual review of the literature related to the cohort’s disease. For other genes, thresholds were adjusted to ensure they remained below the frequency of the most prevalent known PLP variants for each gene, as classified in ClinVar. For example, after an initial review of literature, we determined the PM2 threshold of RP to be 1:4000 or 0.00025. The most common known PLP variant in the RP1 gene in ClinVar associated with IRD is NM_006269.2:c.6181del, which has an East Asian allele frequency of 0.000192. Therefore, the PM2 threshold for candidate variants in this gene was lowered to this frequency. Variants would be designated as supporting PM2 (PM2_Supporting) if their population allele frequencies in gnomAD fall below the derived gene-specific threshold.
PS4: To evaluate the increase in variant prevalence among patients, two distinct sources were utilized: (1) the number of submissions for each variant in ClinVar, categorized as pathogenic, likely pathogenic, likely benign, and benign, were extracted from the most recent ClinVar FTP site; (2) the prevalence of each allele within the same cohort was quantified. Occurrences in patients were further evaluated using threshold-based rules adopted from several gene-specific ClinGen VCEP guidelines, Glaucoma VCEP and Hearing Loss VCEP. These thresholds were applied alongside the PM2 criterion to assign PS4 levels.
PS1/PM5: To determine if the same or different amino acid change had been reported as pathogenic, variants reported a high (≥ 2) gold star PLP classification in ClinVar, including those vetted by the ClinGen-reviewed variant list, were incorporated into a known positive variant repository. This repository was then utilized to ascertain whether the variant under examination exhibits the same or a different amino acid substitution at the identical position within the peptide, compared to the variants in the known positive list.
PM3: To determine if a variant reported in trans with other pathogenic variants, variants located in genes co-occurring with at least one other known positive variant, or a novel PLP variant (defined in S-BCAW Step 2) would be pre-assigned for PM3. The criterion for a known PLP variant was the same as defined in PS1/PM5. According to the ClinGen Sequence Variant Interpretation Recommendation for the in trans Criterion (PM3) (Group 2019), if the concurrent variant in the gene is pathogenic, the PM3_Supporting classification is assigned, indicating a stronger correlation with pathogenic potential. Conversely, if the co-occurring variant is likely pathogenic, a PM3_Tentative classification is assigned, reflecting a lower strength of evidence, quantified as 0.25 points. This does not meet the threshold for full support but provides suggestive evidence for further review. The mode of inheritance (MOI) of the associated disease was considered in Step 2.
Some criteria that were automatically assigned simply apply the threshold-based rules used in MICAW, including PP2 (missense intolerance gene) based on gene specific missense constraint z-score from gnomAD, and PP3 (in-silico evidence) based on REVEL score.
In addition, Mastermind (Chunn et al. 2020), designed for searching scientific literatures by genomic variations, was included in the S-BCAW workflow. The number of scientific publications associated with the variants involved was fetched from Mastermind API, together with the variant specific link to Mastermind website for literature review. This integration helps reviewers to access related ACMG criteria that relies on scientific literatures: PP1 (cosegregation), PS4, PM3 and PP4 (disease specific phenotype).
S-BCAW Step 2: variant classification based on calculated ACMG criteria, gene inheritance mode and variant zygosityAfter the automatic assignment of ACMG criteria, variants were classified into different actionable baskets for further evaluation (Supplementary Table S2). Firstly, combined ACMG points were calculated according to point values for ACMG/AMP strength of criterion categories, designed based on a Bayesian probability model to assess the likelihood of pathogenicity (Tavtigian et al. 2020). Subsequently, the total number of variants present within each gene in the filtered variant list was tallied. Additionally, the gene’s MOI and the zygosity of the variants were considered. Variants were allocated to the PLP classification basket if the pathogenicity assessment, combined with the count of variant(s) in the gene, substantiates a significant correlation with the disease under investigation in the cohort. Variants that contained but did not have sufficient pathogenic ACMG criteria to reach likely pathogenic classification were placed into the Possibly Pathogenic (PossP) basket. The remaining variants, which did not qualify for the categories, were designated as Variants of Uncertain Significance (VUS) and were excluded from the initial automated process to be revisited during subsequent manual curation. This systematic approach ensures a structured and rigorous evaluation of genetic variants, facilitating precise stratification based on their potential clinical relevance.
S-BCAW Step 3: Manual curation for variants in PLP and possible pathogenic basketFor variants allocated to the PLP and PossP classification baskets, the automatically assigned ACMG criteria underwent manual review to corroborate the validity of the sources. Variants in the PLP basket were prioritized to ensure timely and accurate assessments, which was essential for robust disease causative and pathogenicity evaluations. Furthermore, ACMG criteria related to literature evidence and sample phenotypes were meticulously evaluated and additionally assigned by experts. Variants in the VUS basket were not included for manual review.
After this manual intervention, the combined ACMG points and the classification of each variant were updated to reflect the additional insights gained during the review process. This comprehensive approach allowed for a more accurate determination of variant pathogenicity.
Cases containing variants that remained in the PLP basket or promoted from PossP to PLP basket after thorough review were classified as positive cases. Concurrently, the variants in the PLP basket for these positive cases were designated as causative pathogenic (P) or likely pathogenic (LP) variants according to their reviewed ACMG classification. Conversely, samples without PLP variants, or where PLP variants could not be substantiated upon manual review, were classified as negative cases. This classification strategy facilitated a clear distinction between cases likely associated with the disease phenotype and those not linked to the observed clinical manifestations.
Result comparison and reportingDiagnostic outcomes derived from the two methodologies were compared prior to further analysis, encompassing the diagnostic conclusions of the cases, the identification of disease-causing variants, and the assigned ACMG criteria. Any discrepancies observed between the results were thoroughly discussed and resolved through a joint review conducted by the reviewers from both approaches.
Additionally, the processing time for each approach, measured from the point of obtaining the filtered variant list to the completion of variant classification, was recorded and compared to evaluate efficiency.
In cases identified as compound heterozygous, the phasing status was validated using Nanopore sequencing to ensure the accuracy of the genetic interpretation. Following these validations and consultations with the referring clinicians, research reports were prepared and subsequently issued to all patients involved in the study.
This structured approach to comparison and validation ensured that the diagnostic results were both accurate and consistent, providing a reliable foundation for subsequent clinical decision-making.
Comments (0)