
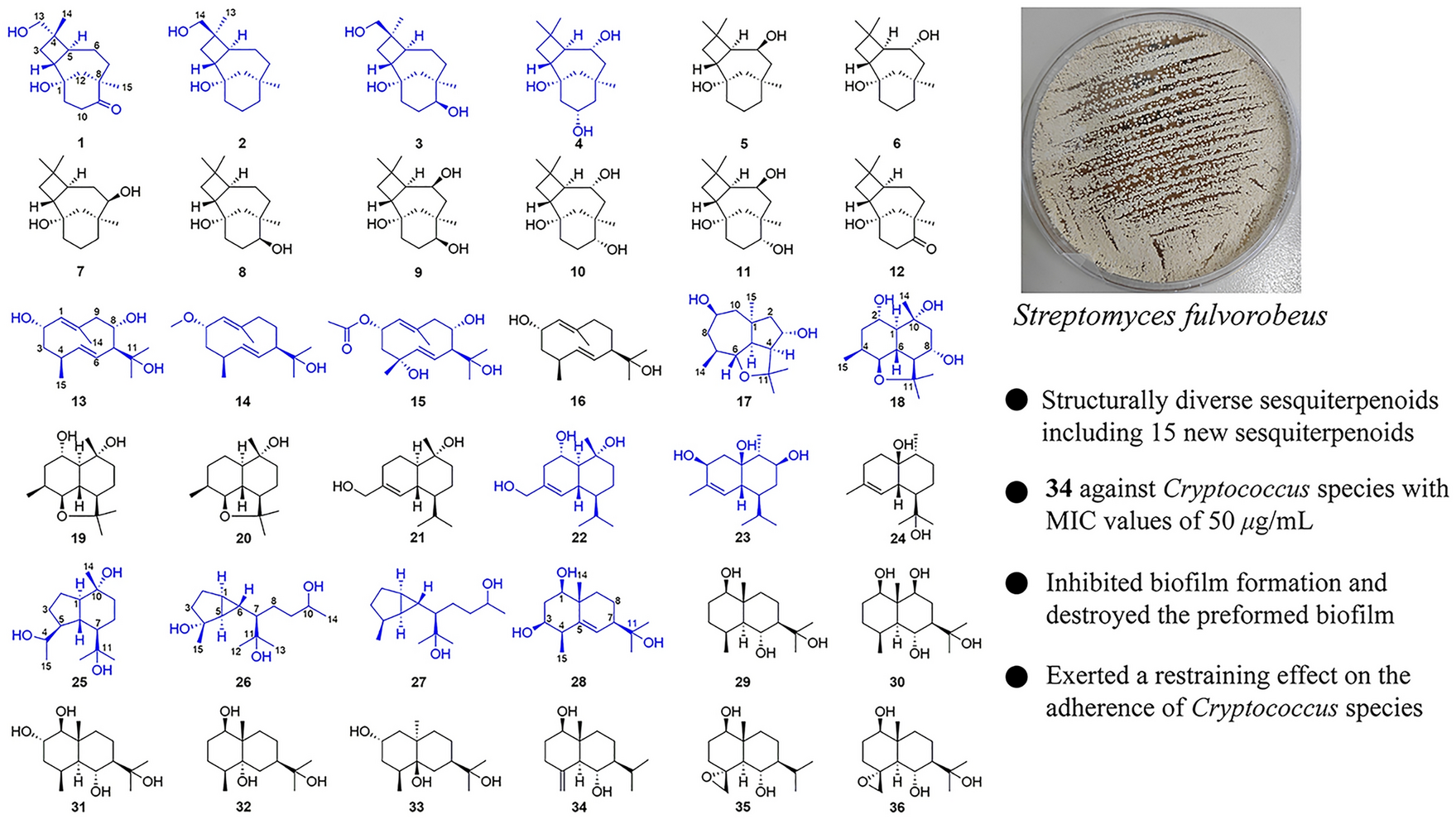
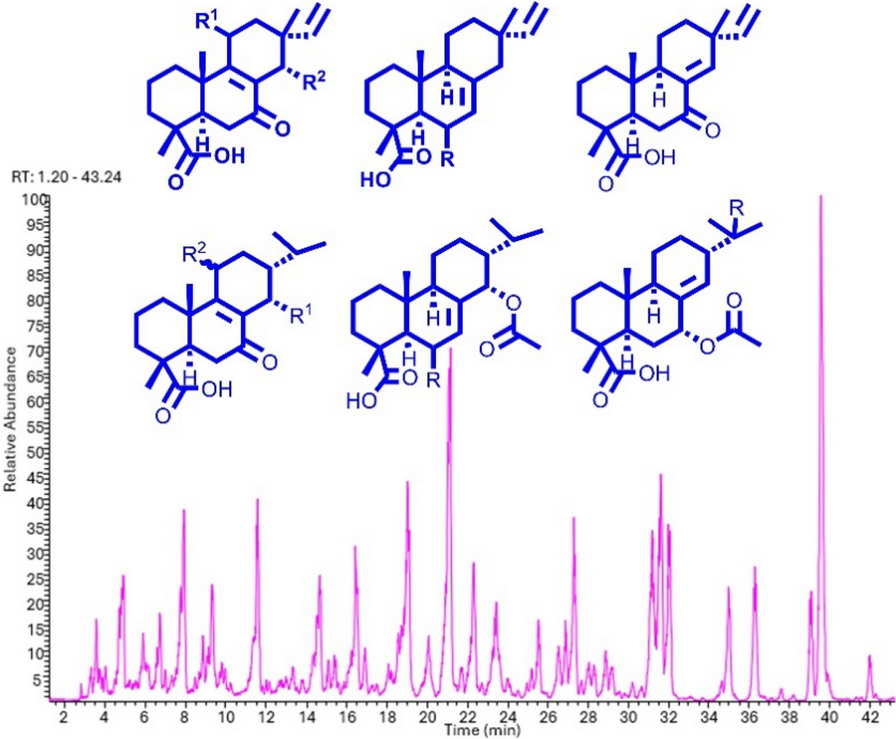
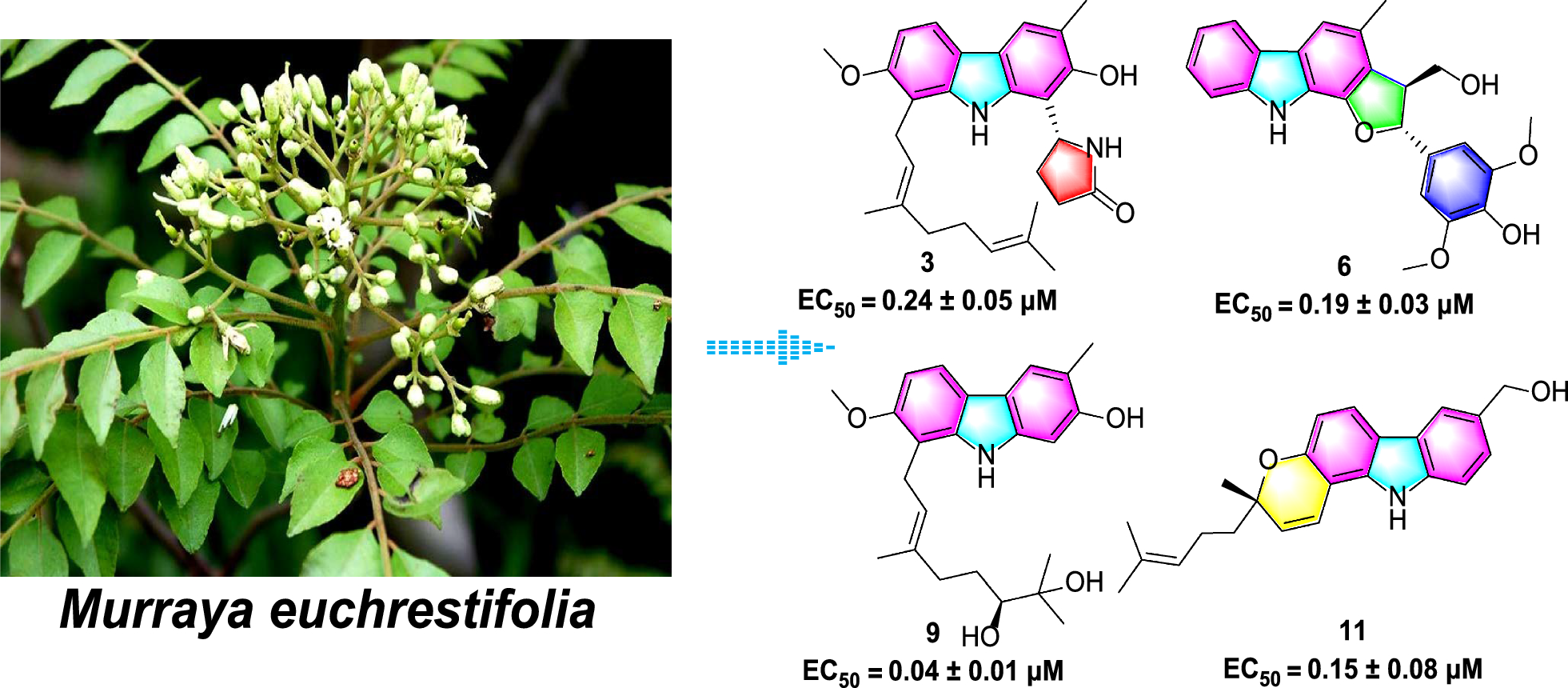
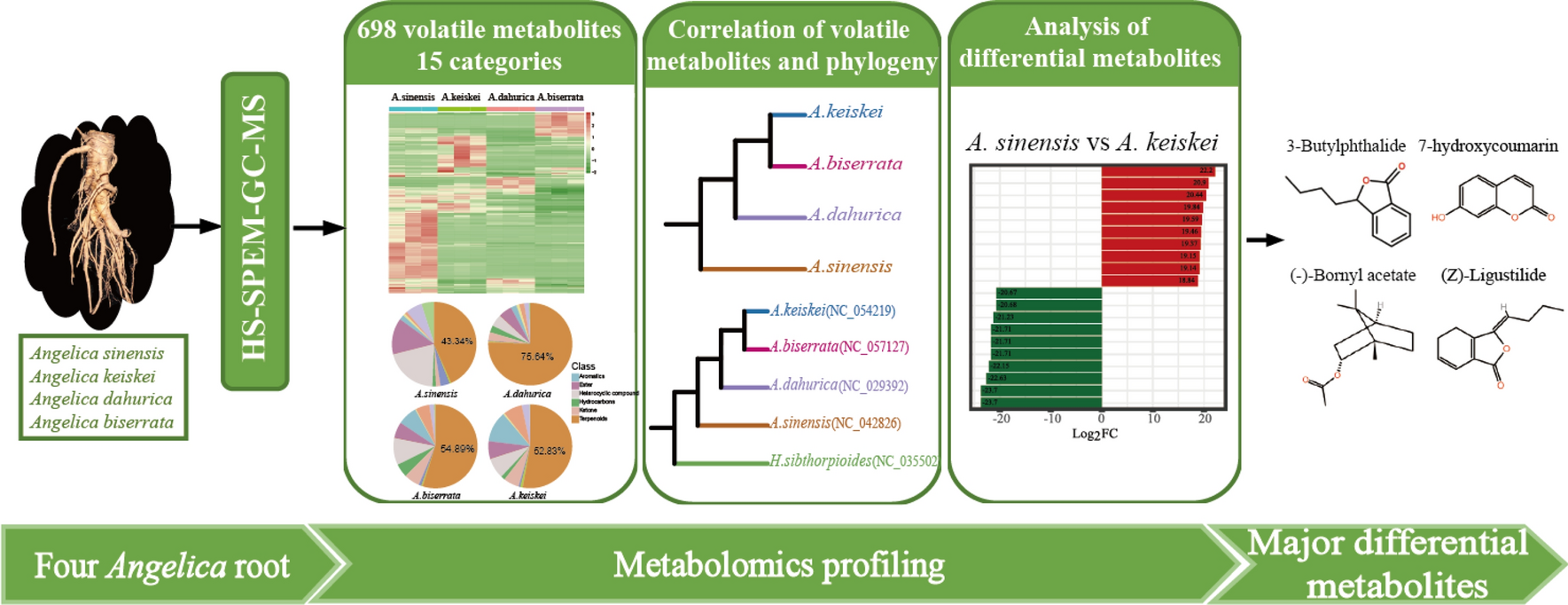
Remember me

Kaemtakol A (1), colorless crystals, has a molecular formula C24H34O7, deduced from the HRESIMS [M + Na]+ at m/z 457.2204 and supported by 13C and DEPT 135 NMR data, suggesting eight unsaturation indices. The IR absorption peaks at 3415 cm–1 and 1740 cm–1 indicated hydroxy and ester functional groups. The 1H- NMR (Table 1) exhibited signals for three quaternary methyls δH/δC 1.09/25.4 (CH3-17), 1.37/22.6 (CH3-18), and 1.08/34.5 (CH3-19); two acetyl methyls at δH 2.12 and 2.22; an oxymethylene group at δH/δC 3.53 and 3.96 (each 1H, d, J = 10.4 Hz)/65.8; two oxymethine groups at δH/δC 4.78/71.9 and 5.64/71.7; a vinyl group with the δH values of 4.98 (1H, d, J = 10.6 Hz), 5.04 (1H, d, J = 17.6 Hz), and 5.86 (1H, dd, J = 17.5, 10.6 Hz); and an olefinic proton at δH/δC 5.95/132.9. The 13C-NMR and DEPT of compound 1 presented twenty-four carbon signals, which matched with five methyl carbons including two acetyl methyls, six methylene carbons including one alkene and one oxygenated, five methine carbons including two oxymethines and two olefinics, and eight quaternary carbons including one alkene, two carbonyls, one oxygenated, and one hemiketal (Table 1). Excluding the above four degrees of unsaturation from two double bonds and two carbonyls, the remaining four unsaturation indices implied a tetracyclic compound. Hence, the structure of 1 was identified as C-20 oxygenated methylene pimarane diterpenoid, featuring a tetracyclic ring bearing two acetoxy groups. The COSY correlations of four spin systems comprising H-1/H2-2/H2-3, H-5/H-6, H2-11/H2-12, and H-15/H2-16, together with the HMBC correlations connecting H3-18 and H3-19 to C-3, C-4, and C-5; H2-20 to C-1, C-5, C-7, and C-9; H-1 to C-3, C-5, and C-20; H-5 to C-6, C-10, C-18, C-19, and C-20; H-14 to C-7, C-9, C-13, C-15, and C-17; and H3-17 to C-12 and C-14, suggested the structural architecture of 1 (Fig. 2). Based on the NOESY experiment, it was indicated that rings A and B displayed a trans-decalin, with the α-axial hydrogen on C-5 and a β-axial position of C-20. Additionally, the acetoxy group on C-1 was found to be in an α-axial position, and the oxygen linkage connecting C-7 and C-20 was situated above the B-ring plane. The C-6 relative configuration was assigned by a large coupling constant value of 3J5,6 (\(\approx\) 10 Hz) which implied that the dihedral angle between them was nearly 0 degree, indicating a cis relationship [12]. This relationship was further proved by the presence of H-5 and H-6 with H3-19 correlations, and the absence of correlation of either H3-18 or H2-20 in the NOESY spectrum (Fig. 2). Thus, these above observations revealed the β-equatorial position of the OAc group on C-6. The X-ray crystallography analysis supported the assigned conformation of 1 and the visual representation of the crystal structure provided by ORTEP drawing is depicted (Fig. 3). Consequently, 1 was structurally determined as 7α,9α-dihydroxy-1α,6β-diacetoxy-7β,20-epoxyisopimara-8(14),15-diene. Furthermore, based on the high resemblance between the calculated and measured ECD spectrum, the (1S,5S,6S,7R,9S,10S,13R) absolute configuration of 1 was determined (Fig. 4).
Table 1 1H (400 MHz) and 13C (100 MHz) NMR spectroscopic data of 1–4 in CDCl3Fig. 2
Key COSY (red dash), HMBC (blue curved arrow), and NOESY (blue dotted curve arrow) correlations of compounds 1–4
Fig. 3
Crystal structure of compound 1
Fig. 4
Simulated and calculated ECD spectra of 1–4
A white amorphous compound 2 named kaemtakol B was formulated as C22H32O6 by HRESIMS at m/z of 415.2099 [M + Na]+. NMR analysis revealed that compounds 1 and 2 were C-20 oxygenated methylene pimarane-type diterpenoids bearing an epoxide ring (Table 1). Their NMR data closely resembled each other, with the only difference being the presence of a secondary alcohol on C-1 in 2. This alcohol at C-1 (δC 67.8) position was confirmed using HMBC cross-peaks that connected H-1 (δH 3.77) to C-5, and H2-20 to C-1 and C-5. Additionally, an oxygen linkage connecting C-7 and C-20 resembling that of 1 could be established from HMBC cross-peaks connecting H-1 and H-5 to C-20; and H-5, H-14, and H2-20 to C-7. The above analyses enabled the establishment of the 2D structure of 2. The NOESY experiment (Fig. 2) unveiled an identical trans-decalin A/B ring pattern to 1, with the hydroxy group on C-1 occupying an α-equatorial position, the 7,20-oxygen bridge situated above the B-ring plane, HO-7 positioned in an α-orientation, and H2-20 in part of the 7,20-oxygen bridge located in a β-orientation. Consequently, 2 was structurally determined as 1α,7α,9α-trihydroxy-6β-acetoxy-7β,20-epoxyisopimara-8(14),15-diene. The high resemblance between the calculated and measured ECD spectra of 2 strongly suggested the (1S, 5S, 6S, 7R, 9S, 10S, 13R) absolute configuration (Fig. 4).
The 1D-NMR spectra of kaemtakol D (3) suggested the presence of a C-20 oxygenated methylene pimarane diterpenoid, and its structure appeared to resemble that of 2 (Table 1). The main differences were detected in the placement of the C-6 hemiketal hydroxy group at δC 107.3 and the C-7 acetoxy group at δC 78.1. These differences were confirmed through HMBC cross-peaks connecting H-5 and H2-20 to C-6, and H-5 and H-14 to C-7 (Fig. 2). NOESY interactions and the optimized structure obtained from the conformer analysis were used to determine compound 3 relative configuration (Fig. 2), which suggested the trans-decalin arrangement of the A/B rings having H-5 positioned in an α-axial orientation and C-20 in an β-axial orientation. As a consequence, the β-oriented acetoxy group at C-7, the α-oriented hydroxy group at C-1, and the oxygen linkage connecting C-6 and C-20 above the B-ring plane were assigned. Hence, compound 3 was determined to be 1α,6α,9α-trihydroxy-7β-acetoxy-6β,20-epoxyisopimara-8(14),15-diene. The high resemblance between the calculated and measured ECD spectra of 3 established the (1S, 5S, 6S, 7R, 9R, 10S, 13R) absolute configuration (Fig. 4).
Kaemtakol D (4) presented a molecular formula C22H28O5, deduced from the HRESIMS ion at m/z 395.1827 [M + Na]+, consistent with nine unsaturation indices. The IR absorption peaks at 3352, 1732, and 1714 cm–1 suggested hydroxy and carbonyl functional groups. The 13C-NMR data revealed twenty-two carbon signals as the resonances of four singlet methyls (one of which being an acetoxy), five methylenes (one of which being an oxymethylene carbon), six methines (including one oxymethine carbon and one sp2 hybridized aldehyde carbon), and seven quaternary carbons (including one olefinic carbon and two sp2 hybridized carbonyls). The abovementioned functional groups attributed to four out of the nine degrees of unsaturation, leaving five degrees to be deduced as a pentacyclic ring. The 1H-NMR spectrum exhibited four methyl protons at δH 2.05 (an acetyl group), 1.32, 1.28, and 1.05, oxymethylene protons at δH 4.83 and 4.59, an oxymethine proton at δH 5.13, as well as an aldehyde proton at δH 9.93 (Table 1). The COSY spin systems led to the assignment of three fragments: H-1/H2-2/H2-3 for C-1 to C-3, H-5/CHO for C-5 with an aldehyde function, and H-14/H-15/H2-16/H-11/H2-12 for C-11 to C-16 (Fig. 2). By deduction from HMBC correlations (Fig. 2), these fragments expanded to form the major portion of the molecules. Furthermore, the HMBC correlations were used to establish the bond connections between rings A and B, whereby the observed cross-peaks from H-5 to C-1 (δC 73.8), C-6 (δC 202.9, an aldehyde group), C-10, C-20 (δC 69.7), and a gem-dimethyl group, from H-6 to C-5, from a gem-dimethyl group to C-3 and C-5, and from H2-20 to C-1, C-5, C-7 (δC 163.7, an ester carbonyl group), C-9 (δC 154.6, an alkene group), and C-10, indicated the appearance of a six-membered carbocycle (A) with a geminal dimethyl group at C-4, an aldehyde unit at C-5, a lactone ring (B), two oxygen atoms (at C-1 and C-20), along with two quaternary carbons (at C-4, and C-10). Therefore, rings A and B are united by a bridged spiro lactone moiety. The incorporation of the remaining tricyclo unit to complete a pentacyclic structure was guided by the HMBC cross-peaks connecting H-11 to C-8 and C-13, H2-12 to C-9, and H-14 to C-9 and C-17 (Fig. 2). In addition, an isolated methylene unit that displayed an AB geminal coupling pattern of H2-16 (δH 1.54 and 0.53) was assigned to be at the junction between C-11 and C-13, and this position was further verified by long-range correlations between H-11 and C-15, H2-16 and C-9, H2-16 and C-12, as well as H3-17 and C-14 and C-15. Moreover, the cross-peaks from H-14 to C-7 (δC 163.7) signified the incorporation of the carbonyl carbon into the lactone ring, thereby necessitating the linkage of C-7 to C-8. The last remaining assignment of carbonyl carbon for the acetate group (OAc, δC 169.4) was positioned at C-1 due to a higher chemical shift value (δC 73.8) in the downfield region, compared with C-1 in compounds 2 and 3 (67.8 ppm). The compound 4 relative stereochemistry was assigned using the NOESY correlations of H3-18/H-6, H3-19/H-5, and H-5/H-3α (Fig. 2). Further evidence for the α-configuration of the C-1 acetoxy group was provided by NOESY cross-peaks among H-1, H-6β, and H2-20, which inferred their co-facial relationship. The location of C-13 was deduced from strong correlations connecting H-12β and H-14 to H3-17β, and the lack of correlation between H-11 and either H-14 or H-15 in the NOESY spectrum. Based on these considerations, 4 was characterized as 1α-acetoxy-13β-methyl-6,7-seco-7,20-olide-8,9-tricyclo[3.2.1.02,7]octane-isopimara-8-en-6-al.
The relative configuration of the carbon positions 5, 10, 11, 13, and 14 in 4 was determined by DP4 + NMR chemical shift probability analysis. 1H and 13C chemical shifts for four possible isomers (4 and 4a–4c) were computed and compared to the experimental values (see Additional file 1). The statistical analysis confirmed the agreement with isomer 4, with a 100% probability. Furthermore, the high resemblance between the calculated and measured ECD spectra of 4 established the (1S,5S,10S,11R,13R,14S,15S) absolute configuration (Fig. 4).
It is noteworthy that compound 4 is partially associated with trachylobane diterpenoids [13, 14], known for their characteristic cyclopropane moiety. However, 4 stands out from this structural family by incorporating two additional functional elements, which are the adjacent tricyclo[3.2.1.02,7]octene ring system [15, 16] and a spirocyclic lactone ring junction at C-10, thereby expanding the diversity of this particular structural type.
The putative biosynthetic pathway of compounds 1–4 is illustrated in Fig. 5. Compounds 1–4 are possibly formed from geranylgeranyl pyrophosphate (GGPP) through the pimara-8(14),15-diene (I). Oxidation at multiple sites of I gives intermediate II, which is presumably a common biosynthetic precursor of compounds 1–4. Further oxidation at C-6 of II followed by ketal formation gives IV, which is then acetylated at 7-OH to give 3. Alternatively, oxidation at C-7 of II followed by ketal formation provides intermediate VI. Acetylation at different sites of VI yields compounds 1 and 2 and intermediate VII. Compound 4 is proposed to originate from VII, whereby the glycol cleavage followed by dehydration generates the diene moiety in IX. Upon intramolecular Diels–Alder reaction with the adjacent double bond in IX, compound 4 is formed.
Fig. 5
Putative Biosynthetic Pathways to Generate 1–4
Kaemtakols A–D (1–4) were screened for their inhibitory activity in suppressing the NO production triggered by lipopolysaccharide in RAW 264.7 macrophage cells and the NF-κB production activated by cytokine-induced inflammation in HaCat human skin cells. The results (Table 2) showed that 2 potently inhibited NO production having an IC50 of 0.69 μM. In contrast, substituting the acetylated group at position 1 of compound 2, namely kaemtakol A (1), abolished anti-inflammation activity (IC50 = 103.9 μM). In addition, compounds 1–3 satisfyingly exhibited minimal cytotoxicity towards RAW 264.7 cells at the specific concentrations needed to inhibit NO production. Nevertheless, these compounds did not show discernible inhibitory effects against NF-κB (EC50 > 100 μM).
Table 2 Inhibitory effects of the compounds 1–4 against the LPS-induced NO production in RAW264.7 cells and NF-κB production by cytokine induced inflammation in HaCat human skin cellsTo determine the possible binding mode of compound 2 to iNOS, docking studies were conducted using the crystal structure with high-resolution iNOS protein (PDB 3E6T). Results (Fig. 6a, b) revealed that the binding position of 2 was next to cofactors: HEM-901 and H4B-902, and overlapped partly with that of the iNOS inhibitor AR-C118901 [17]. Compound 2 exhibited a binding energy of − 8.0 kcal/mol, which was marginally greater than that of AR-C118901 (− 9.2 kcal/mol), indicating that 2 and the cocrystal inhibitor had roughly equivalent binding affinities (Table S16). Moreover, 2 occupied the binding pocket through hydrophobic interactions and hydrogen bonds with crucial amino acid residues, including GLN257, ARG260, TYR341 and ARG260 (Fig. 6c). As depicted in Fig. 6d, the hydroxy and acetoxy groups of 2 played an important role in hydrogen contacts with GLN257 and ARG260, while its alkene and methyl groups took part in the hydrophobic interactions with MET114, TRP84, TYR341, and TYR367 in the cavity of iNOS.
Fig. 6
The binding position of compound 2 (pink) in a the whole receptor containing AR-C118901 (green) and cofactor: HEM-901 and H4B-902 (blue), and b the binding pocket of iNOS. c Potential hydrogen contacts (green dash) and hydrophobic interactions (pink dash) of compound 2 with amino acid residues in the binding site of iNOS. d 2D diagram representing interactions of compound 2 in the cavity of iNOS (PDB ID: 3E6T)
Comments (0)