
Remember me
Our research complies with all relevant ethical regulations and guidelines. The lung fibrosis experiments were performed in compliance with guidelines established by the Barcelona Science Park’s Committee on Ethics for Animal Experimentation (CEEA) and under approved protocol no. 10884. All other mouse procedures were performed under licence, according to the UK Home Office Animals (Scientific Procedures) Act 1986, ARRIVE and local institutional guidelines. The mouse pituitary experiments were approved by the UCL ethical review committee (PPL P5FB9D417). Liver cancer initiation and the WD experiments were approved by the animal welfare and ethical review board at Imperial College London (PPL 70/09080 and PPL PE02064666, respectively). Cancer xenograft experiments were performed by national and international guidelines and were approved by the institutional review board at Southampton University (PPL P81E129B7).
DrugsThe following compounds were used in the present study: ABT-263 (Selleckchem, S1001), etoposide (Sigma-Aldrich, E1383), QVD-OPh hydrate (Sigma-Aldrich, SML0063), 4OHT (Sigma-Aldrich, H7904), doxycycline hyclate (Sigma-Aldrich, D9891), doxorubicin hydrochloride (Cayman Chemical, 15007), triamcinolone (Selleckchem, S1933), beclomethasone dipropionate (Selleckchem, S3078), GSK2606414 (Tocris, 5107), GSK2656157 (Selleckchem, S7033), GCA (Selleckchem, S7266), BFA (Selleckchem, S7046), IMP1088 (Myricx), DDD86481 (Myricx), IMP1320 (Myricx) and bleomycin sulfate (Generon, A10152).
AntibodiesThe following primary antibodies were used in this study: mouse monoclonal anti-BrdU (3D4, BD Biosciences, 555627) 1:2,000, mouse monoclonal anti-p16INK4a (JC8, CRUK) 1:1,000, rabbit polyclonal anti-glyceraldehyde 3-phosphate dehydrogenase (anti-GAPDH) (Abcam, ab22555) 1:2,000, mouse monoclonal anti-IL-8 (6217, R&D systems, MAB208) 1:100, goat polyclonal anti-IL-6 (R&D Systems, AF-206-NA) 1:40–1:200, mouse monoclonal anti-ARF1/3/5/6 (1D9, Invitrogen, MA3-060) 1:500, rabbit monoclonal anti-COPB2 (899, gifted from F. Weiland) 1:10,000, mouse monoclonal anti-EEA1 (14, BD Biosciences, 610457) 1:200, rabbit polyclonal anti-XBP1 (Abcam, ab37152) 1:200, rabbit polyclonal anti-ATF6 (Abcam, ab37149) 1:500, sheep polyclonal anti-TGN46 (Bio-Rad, AHP500G) 1:400, mouse monoclonal anti-GM130 (35, BD Biosciences, 610822) 1:500, mouse monoclonal anti-CHOP (L63F7, CST, 2895S) 1:1,000, rabbit monoclonal anti-p21CIP1 (12D1, CST, 2947S) 1:2,000, rabbit monoclonal anti-p21CIP1 (EPR18021, Abcam, ab188224) 1:700, mouse monoclonal anti-N-Ras (F155, Santa Cruz, sc-31) 1:100, mouse monoclonal anti-β-catenin (6F9, Sigma, C7082) 1:500, rabbit polyclonal anti-β-catenin (Thermo, RB-9035-P1) 1:500, mouse monoclonal anti-synaptophysin (27G12, Leica, SYNAP-299-L) 1:200, rabbit polyclonal anti-CC3 (CST, 9661S) 1:1,000, goat polyclonal anti-CXCL1 (R&D, AF-275) 1:100, mouse monoclonal anti-BMP2/4 (100230, R&D, MAB3552), mouse monoclonal anti-VEGF (23410, R&D MAB2931) 1:100, mouse monoclonal anti-GM-CSF (3209, R&D, MAB215) 1:100, rabbit polyclonal anti-CD68 (Abcam, ab125212) 1:100, rabbit polyclonal anti-ARF1 (10790-1-AP, Proteintech) 1:1,000, rabbit polyclonal anti-ARL1 (16012-1-AP, Proteintech) 1:1,000, rabbit polyclonal anti-PPM1B (HPA-016745, Cambridge Bioscience) 1:1,000 and rabbit polyclonal, mouse monoclonal anti-TUBA (ab1729, Abcam) 1:1,000.
We used the following secondary antibodies: goat anti-mouse IgG-HRP (immunoglobulin-G–horseradish peroxidase; Santa Cruz, sc-2005) 1:2,000, goat anti-rabbit IgG-HRP (Santa Cruz, sc-2004) 1:2,000, goat anti-mouse IgG (H + L) AlexaFluor488 conjugated (Invitrogen, A-11029) 1:2,000, goat anti-mouse IgG (H + L) AlexaFluor594 conjugated (Invitrogen, A-11032) 1:2,000, goat anti-rabbit IgG (H + L) AlexaFluor594 conjugated (Invitrogen, A-11037) 1:2,000, donkey anti-sheep IgG (H + L) AlexaFluor594 conjugated (Invitrogen, A-11016) 1:2,000 and donkey anti-sheep IgG (H + L) AlexaFluor488 conjugated (Invitrogen, A-11015) 1:2,000. For the IpaJ western blot experiments, we used the following secondary antibodies: IRDye 800CW goat anti-rabbit IgG (H + L, 926-32211, Li-Cor, 1:10,000)and IRDye 800CV goat anti-mouse IgG (H + L, 926-32210, Li-Cor, 1:10,000).
Cell linesIMR90 (ATCC, CCL-186), SK-HEP-1 (ATCC, HTB-52), A549 (ATCC, CCL-185), HFFF2 (ECACC, 86031405), HCT116 (ATCC, CCL-247), MCF7 (ATCC, HTB-22), 5PT39 (a gift from I. C. Mackenzie, QMUL), PBEC (ATCC, PCS-300-010) and NHLF (Lonza, CC-2512). PBECs were cultured in airway epithelial cell basal medium (ATCC-PCS-300-030; ATCC) supplemented with bronchial epithelial cell growth kit supplements (ATCC-PCS-300-040; ATCC) and 0.1% antibiotic–antimycotic solution (Gibco) with media replenished every 48 h. Adult NHLFs were cultured in fibroblast basal medium (CC-3131; Lonza) supplemented with a SingleQuot Kit of supplements and growth factors (CC-4126; Lonza), with media replenished every three to four days as required. All other cells lines were maintained on Dulbecco’s modified eagle medium (DMEM; Gibco) supplemented 1% 100X Gibco antimycotic-antibiotic and 10% (vol/vol) FBS (Labtech, Batch 41213), hereinafter referred to as DM10 medium. Passaging of cells was performed by enzymatic detachment using 0.05% Trypsin-EDTA (Gibco) on cells for 5 min, followed by inactivation in DM10 medium and centrifugation at 180g for 5 min. The supernatant was aspirated to remove dead cells and debris, and the pellet was resuspended in fresh DM10. Cell numbers were determined using a Guava EasyCyte platform (Millipore) using Guava ViaCount reagent. In-built GuavaSoft software was used to define live cells and remove cell debris/dead cells from the final cell count. Experiments using IMR90 cells or cell lines generated from them were carried out using cells between passages 10 and 14. To generate ER:RASGV12 and other derived cells, IMR90 or HFFF2 cells, retroviral and lentiviral infections were carried out as described in ref. 32. Treatment with 100 nM 4-OHT (Sigma, in DMSO) was used to induce IMR90 ER:RAS cells to undergo OIS. Therapy-induced senescence (TIS) was induced in IMR90 cells by treatment with 33 μM (50 µg ml−1) bleomycin sulfate (Generon, A10152) for 24 h, 20 μM palbociclib (Selleckchem, S1116) for seven days or 100 nM doxorubicin (Cayman Chemical, #15007) for seven days. Senescence was induced in A549 and SK-HEP-1 cells by treatment with 2 μM etoposide (Sigma-Aldrich, E1383) for seven days. HCT116 senescence was induced by 100 nM treatment with doxorubicin (Cayman Chemical, 15007) or 2 µM etoposide for three days, followed by four days culture in medium without chemotherapy. Senescence was induced in MCF7 by treatment with 200 nM doxorubicin or 2 µM etoposide for seven days.
MiceAll mice were purchased from Charles River UK Ltd except where noted otherwise.
For HDTVI experiments, female C57BL/6J mice aged five to six weeks were given 20 μg of a vector expressing NrasG12V and Gaussia luciferase (Gluc) along with 5 μg of SB13 transposase-expressing plasmid. Experiments were performed as described in ref. 55. Four days after HDTVI, mice were bled to assess the presence of a Gaussia luciferase signal in the blood plasma and used to randomize groupings for vehicle and drug-treated groups. On day 5, mice were given 25 mg kg−1 of IMP1320 (n = 9 mice) or vehicle (n = 9 mice) (10 mM Na2HPO4-7H2O and NaH2PO4H2O buffer, 0.2% Tween-80, pH 7.4) intraperitoneally (i.p.) daily for four days. Twenty-four hours after the last drug injection, mice were culled and livers collected for paraffin embedding and frozen in optimal cutting temperature compound (OCT).
For cancer xenograft experiments, 6.7 × 105 5PT cells ± 2 × 106 HFFF2 cells were injected subcutaneously (s.c.) into the flanks of immunocompromised, male NOD SCID Gamma (NSG) mice (three to five months old). For knockdown experiments, HFFF2 fibroblasts expressing inducible shRNAs targeting COPA, COPB2 or control were irradiated at 10 Gy using a MultiRad350 X-ray irradiation cabinet (from Precision X-ray) just before implantation. In vivo, expression of the shRNA was induced using doxycycline given in drinking water throughout the experiment (2 mg ml−1 with 5% sucrose in the drinking water). Experiment A (Fig. 5e) included six mice for shCOPB2.1 + irradiation and seven mice for all other groups. In experiment B (Supplementary Fig. 12f) n = 7 mice per group were utilized. The data presented in Extended Data Fig. 5d compile all mice used in experiment A and experiment B. For the control experiment described in Extended Data Fig. 5e, n = 6 mice were included in the 5PT group and n = 8 mice in all other groups.
For xenograft experiments with NMTi (Fig. 7e and Extended Data Fig. 7f), 10 mg kg−1 NMTi (DDD86481) was dissolved in water containing 5% DMSO, 20% PEG400, 10 mM Na2HPO4.7H2O and NaHPO4H20 buffer, 0.5% Tween-80, pH 7.3, and administered by intraperitoneal (i.p.) injection as indicated in Fig. 7e. Tumour size was measured over time using an electronic caliper and calculated using the formula 4π/3 × r3 (radius (r) calculated from the average diameter, measured as the tumour width and length). For this experiment, n = 6 mice were utilized for the 5PT + vehicle treatment group and n = 8 mice per group for all other groups. We did not exceed the maximal tumour size permitted under the licence (1,750 mm3) during the experiments. The area under the curve (AUC) for each tumour within a treatment group for single experiments was analysed, and statistical analysis comparing the AUCs was performed on pooling multiple experiments.
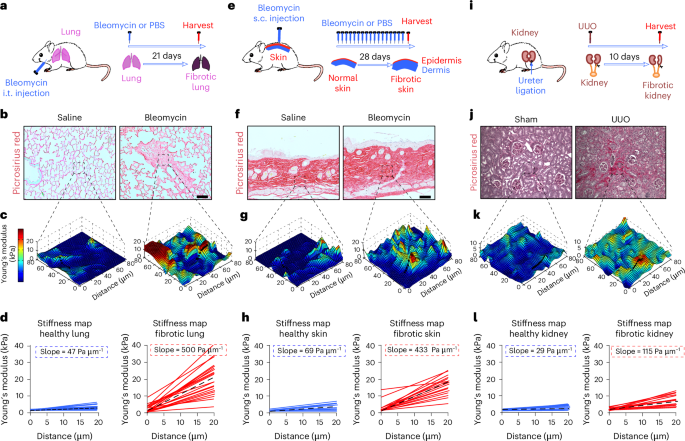
For testing senolytics ex vivo in the ACP model of OIS, neoplastic pituitaries from 18.5dpc Hesx1Cre/+;Ctnnb1lox(ex3)/+ embryos67 were dissected. Both male and female embryos were used, and the numbers were equalized in experimental and control groups. For the experiments shown in Fig. 7h,i and Extended Data Fig. 7h, n = 8 18dpc embryos were dissected (n = 5 embryonic pituitaries were treated with vehicle and n = 3 with IMP1088). For the experiments described in Extended Data Fig. 7f,g, n = 14 18dpc embryos were dissected (n = 5 embryonic pituitaries were fixed at t = 0 h; n = 3 embryonic pituitaries were fixed at t = 24 h; n = 3 embryonic pituitaries were fixed at t = 48 h; n = 3 embryonic pituitaries were fixed at t = 72 h). In both sets of experiments, after treatment and fixation, pituitaries were sectioned and stained (the specific numbers of sections stained and analysed for each experiment are described in the figure legends). For the lung fibrosis experiments, we used a previously described mouse model of lung fibrosis induced by intratracheal administration of senescent human cells16,40. Normal proliferating (IMR90 vector) or gamma-irradiated senescent human fibroblasts IMR90 (IMR90 vector, IMR90 shCOPB2.1 or IMR90 shCOPB2.2) (500,000 cells) were delivered into the lungs of six- to eight-week-old athymic (nu/nu) male mice (Envigo Laboratory). Two days before intratracheal instillation, these animals started treatment with doxycycline (1 mg ml−1 in the drinking water) until the end of the experiment. Three weeks after intratracheal instillation, their lungs were removed and analysed. To estimate the number of senescent IMR90 cells engrafted in the lung after 48 h post-instillation, we first performed a calibration using a known amount of IMR90 cells mixed with lung homogenates. Specifically, the right lobes of nude mice were surgically dissected and placed into 1.5-ml tubes. Homogenates of the lung samples were performed by grinding the frozen samples with liquid nitrogen using a mortar and pestle. Tissues were then thawed, 1 ml of distilled water was added to the tissues, and the resulting suspensions were homogenized using a micro-sample homogenizer (Precellys). Different quantities of senescent shControl IMR90 cells (0, 1,000, 5,000, 10,000, 50,000 or 100,000 cells) were mixed with 1 ml of homogenized lung tissue. After Trizol extraction of RNA and cDNA synthesis using SuperScript III reverse transcriptase (Thermo Fisher), real-time qPCR was performed using the PowerUp SYBR Green Master Mix (Applied Biosystems). Gene expression analysis was performed using predesigned primers and probes for human MMP3. Data were normalized using mouse Actin b. The resulting calibration curve was used to interpolate the data obtained using lung samples 48 h post-intratracheal instillation of shControl, shCOPB.1 and shCOPB.2 IMR90 cells (500,000 cells) in nude mice under doxycycline. For lung fibrosis experiment, n = 5 mice per group were used, and for validation of engraftment, n = 3 mice per group were utilized.
For the mouse model of bleomycin-induced lung fibrosis, pulmonary fibrosis was initiated by intratracheal instillation of bleomycin (0.75 U kg−1) into the lungs of six- to eight-week-old male C57BL/6J mice (Envigo Laboratory). Fourteen days after intratracheal instillation, once the mice had developed well-established pulmonary fibrosis, these animals started treatment with NMTi (IMP1320; 10 mg kg−1, i.p.) or vehicle, administered in a cyclical pattern, consisting of three consecutive days of treatment followed by three consecutive days without treatment. Two weeks after treatment started, the lungs were removed and analysed. For these experiments, we used n = 8 mice per group. Two mice in the vehicle-treated group died before the end of the experiment and were not included in any analysis.
For WD experiments, C57BL/6J male mice aged eight weeks (n = 15 for each of the three groups) were placed on chow (4.25% fat, RM3, Special Diet Services) or WD (Research Diets, D16022301; 40% kcal fat (non-trans-fat Primex shortening), 22% (wt:wt) fructose, 10% (wt:wt) sucrose and 2% (wt:wt) cholesterol) for four weeks before the first round of injections. Mice were then injected i.p. daily with vehicle (10 mM Na2HPO4.7H2O and NaH2PO4H2O buffer, 0.2% Tween-80, pH 7.4) or 10 mg kg−1 DDD86481 (5% DMSO, 20% PEG400, 10 mM Na2HPO4.7H2O) and NaH2PO4-H2O buffer, 0.5% Tween-80, pH 7.3, dissolved by cold water bath sonication, for three days, then given two rounds of a four-week rest period and three-day daily i.p. injection. Blood was collected before the final round of injection for physiological assessments. Mice were allowed to rest for four weeks before being culled and organs were collected for freezing in OCT, paraffin embedding, blood collection for physiological measurements, and tissue snap-freezing for RNA extraction. A mouse of the WD + vehicle group died before the end of the experiment and was not included in any analysis.
To assess the effect of NMTi on metabolic function, a cohort of C57BL/6J male mice aged eight weeks were treated with either vehicle (n = 5 mice), 10 mg kg−1 DDD86481 (n = 5 mice) or 25 mg kg−1 IMP1320 (n = 4 mice), then blood was collected after seven days Supplementary Fig. 16a,b). To assess the effect of NMTi on insulin secretion (Supplementary Fig. 16c), 5 µl of blood was collected after seven days from mice treated with either vehicle (n = 5 mice) or 10 mg kg−1 DDD86481 (n = 6 mice). Blood was processed using an Ultra Sensitive Mouse Insulin ELISA kit (Crystal Chem) according to the manufacturer’s instructions. To assess the effect of NMTi on immune cell composition, C57/BL6 mice were treated with either vehicle, 10 mg kg−1 DDD86481 or 25 mg kg−1 IMP1320, and blood was collected one day or seven days after treatment (day 1, n = 5 mice per group; day 7, n = 3 mice per group).
Vector constructionpLNC-ER:RAS-neo has been described previously in ref. 68. The mutant GBF1M832L construct was a gift from F. J. M. van Kuppeveld (Utrecht University). Cloning of GBF1M832L intro retroviral expression vector (pBabe-puro) was performed by PCR amplification using Human5SnaBIGBF1 (5′-CGTACGTAGCCATGGTGGATAAGAATATTT-3′) and Human3SalIGBF1 (5′-CGGTCGACGCCTTAGTTGACCTCAGAGGTG-3′) primers with Q5 High-Fidelity DNA polymerase (New England Biosciences) according to the manufacturer’s instructions. Amplified GBF1M832L was subcloned into pBabe-puro using standard cloning with SnaBI and SalI restriction enzymes. Gaussia luciferase (Gluc) containing plasmid was a gift from U. Griesenbach (Imperial College London). To generate Gluc expressing HDTVI construct (CaNiGluc), Gluc was PCR-amplified using 5BmgBIsogLUX: (5′-GATTAAGACG TGGTTTTCCT TTGAAAAACA CGATGATAAT ATGGGAGTGA AGGTGCTGTT-3′) and 3sogLUXAge1 (5′-TTTGTTACCG GTCTCATCAA TCTCCCCCAGCT-3′) primers and Q5 High-Fidelity DNA polymerase (New England Biosciences), according to the manufacturer’s instructions. Amplified Gluc was subcloned into HDTVI construct (CaNiG) by restriction enzyme excision of GFP and annealing of Gluc amplicon processed with BmgBI and AgeI into CaNiG plasmid. The IpaJ construct was a gift from E. Tate (Imperial College London). Cloning of IpaJWT/C64A was performed by PCR amplification of IpaJ using 5′EcoRIKozIpaJ (5′-tggtggaattcgccaccATGTCGGAACAACGGAAG-3′) and 3′IpaJPmeI (5′-agcaggtttaaacTTACAAAGCCTCATTAGT-3′) and subcloning into pLenti-puro vector (Addgene 39481) with EcoRI and PmeI restriction enzymes. Tetracycline inducible (Tet-ON all-in-one) shRNA vector (LT3GEPIR) was a gift from J. Zuber (IMP, Vienna). The generation of miRE-based inducible shRNA vectors was performed as previously described32. The shRNA sequences used in this study are described in Supplementary Table 1.
IF and high-throughput microscopyIF staining was carried out by first fixing wells of 96-well plates at the desired timepoint for 1 h using 4% paraformaldehyde (PFA; wt/vol, in phosphate-buffered saline (PBS)) followed by washing three times with PBS. Wells were then permeabilized using 0.2% Triton X-100 (vol/vol, PBS) for 10 min and then washed twice with PBS to halt permeabilization. Non-specific antibody binding was blocked by incubation with a blocking solution for 1 h at room temperature (r.t.). The blocking solution contained 1% bovine serum albumin (BSA; wt/vol, PBS) supplemented with 0.4% fish skin gelatin (vol/vol, PBS). Primary antibodies were diluted in blocking solution and wells were incubated with primary antibody solution for 1 h at r.t. For BrdU staining, primary antibody solution was supplemented with 0.5 U μl−1 DNase (Sigma) and 1 mM MgCl2, and the incubation times were reduced to 30 min. Following incubation, the primary antibody was then removed by washing three times with PBS. Secondary antibodies conjugated to Alexa-594 or Alexa-488 fluorophores were then diluted in blocking solution and added to wells to be incubated in the dark for 1 h. The secondary antibody was then removed by washing three times with PBS and nuclei counterstaining with 1 μg ml−1 4′,6-diamidino-2-phenylindole (DAPI; wt/vol, PBS) for 10 min. Wells were then washed with PBS three times.
Immunofluorescence image acquisition was performed using an automated InCell Analyzer 2000 high-throughput microscope. Multiple 96-well plates were placed into stacks by a KiNEDx robotic arm (PAA) running Overlord software so that the plates could be sequentially loaded into, imaged and removed from the InCell microscope. Wells were imaged using a ×20 objective except for wells stained only with DAPI or Golgi-related staining, which were performed at ×10 and 40, respectively, then 2 × 2 binning of images was used to reduce the image file sizes. Fluorophores were imaged using pre-set ‘DAPI’, ‘Texas Red’ and ‘FITC’ wavelengths on the microscope for DAPI stain, AlexaFluor594 and AlexaFluor488, respectively. Eight, 24 and 18 fields per well were captured for the ×10, ×20 and ×40 objectives, respectively.
High-content image analysis was carried out using the InCell Investigator 2.7.3 software (GE Healthcare). DAPI nuclear counterstain was used to segment cells using a top-hat method and used to provide a mask for nuclear-localized stains. For cytoplasmic stains, a 6-μm collar was applied around the cell and, for detection of cytoplasmic organelles such as Golgi, a ‘region growing’ collar was used. Quantification for nuclear staining was measured as the average pixel intensity (greyscale) for the wavelength of fluorophore across the area of the nuclear mask. Cytoplasmic staining quantification was of either the average pixel intensity or the coefficient of variance of pixel intensities within the collar area. Golgi structural analysis utilized a multiscale top-hat segmentation method to detect organelle structures between 1 and 3 pixels in size within a region growing collar. Cells with >25 Golgi organelle structures per cell were classified as cells with dispersed Golgi.
Growth assaysBrdU incorporation and colony formation assays were performed as previously described in ref. 38. Briefly, for BrdU incorporation assays, cells were incubated with 10 μM BrdU for 18 h before being fixed using 4% PFA (vol/vol, PBS). BrdU incorporation was assessed via IF and high-content analysis. For crystal violet staining, cells were seeded at low density in 10-cm dishes and cultured for 10–14 days or until proliferating cells had reached 80–90% confluency. To assess senolysis, cells were seeded in 10-cm plates at high density. Senolytic drugs were added at their indicated concentration in DMSO (<0.5% vol/vol final concentration) and cultured for a further three days. If longer drug treatment was required, fresh drug and media were added on day 3 and cultured for a further four days. At the endpoint, plates were fixed with 0.5% (wt/vol, PBS) glutaraldehyde (Sigma) for 1 h, washed twice with dH2O, and left to dry overnight. Dried plates were then stained with a 0.2% (wt/vol, PBS) solution of crystal violet (Sigma, C6158).
Senolytic assaysSenolytic assays were performed as described previously15. Briefly, at the indicated timepoints, confluent senescent or control cells in 96-well plates were switched to DMEM 0.5% FBS and drugs in DMSO were added (<5% vol/vol final concentration). Drugs were replenished after three days if the assay length was longer than 72 h. For TIS of PBECs (ATCC-PCS-300-010), cells were seeded at passage 3 and treated with bleomycin (100 ng ml−1) or vehicle for five days, followed by washout. Seven days post senescence induction, cells were treated with the indicated drugs for 72 h. Adult NHLFs (Lonza CC-2512) at passages 4 to 5 were seeded into 96-well plates and induced to senesce by treatment with bleomycin (50 mg ml−1), or vehicle, for 24 h. Seven days post-induction of senescence, cells were treated with the indicated drug concentrations for 72 h. Cells were fixed and stained with DAPI, followed by assessment by automated microscopy. The percentage survival was calculated by dividing the number of cells post-drug treatment by the corresponding number of cells treated with the vehicle at the same time.
For senolytics assays during replicative senescence, PBECs were serially passaged until passages 4–6, whereby a mixed population of senescent and growing cells can be distinguished. PBECs were plated into 96-well plates and treated with the indicated drug for 72 h. Cells were fixed and stained with anti-p16 antibody and DAPI, followed by assessment by automated microscopy. The percentage survival for p16-negative and p16-positive fractions was calculated by dividing the number of cells post-drug treatment by the number of cells treated with the vehicle.
Tissue processingOrgans were fixed in 4% PFA overnight before being transferred to 70% ethanol. Tissue processing before paraffin embedding was performed on a Sakura Tissue-Tek VIP 6 automated tissue processor. Briefly, specimens in embedding cassettes were dehydrated by progressing through steps of 70% ethanol for 45 min at 37 °C, 80% ethanol for 45 min at 37 °C, 90% ethanol for 30 min at 37 °C, 96% ethanol for 45 min at 37 °C, 100% ethanol for 30 min at 37 °C, 100% ethanol for 1 h at 37 °C and 100% ethanol for 1 h at 37 °C. Dehydrated samples were then cleared by three washes in xylene for 30 min, 45 min and 1 h at 37 °C. Finally, the specimens were infiltrated by two immersions in 62 °C paraffin wax for 45 min and 1 h, followed by two immersions in 62 °C paraffin wax for 30 min. The specimen was then embedded in a paraffin block on an embedding centre (Leica EG1160), and 4-μm sections were made using a Thermo Fisher scientific microtome (Microm HM355S) and attached to slides.
IHC stainingThe slides were deparaffinized by washing them twice in Histoclear for 5 min each, followed by 5-min washes in decreasing concentrations of ethanol (100%, 75%, 50% and 25% ethanol) before a final wash of 5 min in dH2O. Heat-induced epitope retrieval (HIER) was then performed in a pressure cooker for 20 min using either antigen-unmasking solution, citrate-based at pH 6.0 (VectorLab, H-3300-250), or antigen-unmasking solution, Tris-based at pH 9.0 (VectorLab, H-3301-250), depending on the antibody manufacturer’s instructions. Following HIER, slides were cooled on ice for 10 min and then washed in PBS for 5 min. For intracellular stains, sections were permeabilized with 0.2% Triton X-100 in PBS for 10 min and washed twice in PBS for 5 min. For NRAS staining, liver slides were washed in 0.1% H2O2 in PBS for 15 min, followed by washing twice in PBS to reduce endogenous peroxide activity. Sections were marked using a hydrophobic pen, and non-specific antigen binding was blocked by incubating the slides with CAS-Block histochemical reagent (Thermo Fisher, 008120) for 30–45 min in a humidified chamber. The slides were then incubated with primary antibody overnight in a humidified chamber at 4 °C. Slides were washed twice in PBS for 5 min and incubated with secondary antibody SignalStain Boost IHC detection reagent with mouse HRP (Cell Signalling Technology, 8125) or rabbit HRP (Cell Signalling Technology, 8114) for 30–45 min. Next, the slides were washed twice in PBS for 5 min and incubated for 2–10 min with a SignalStain DAB substrate kit (CST, 8059) to detect the HRP signal. Signal development was stopped when visible positive cells could be detected on a microscope, by washing slides in dH2O. To counterstain the DAB signal, slides were incubated for 30 s in modified Mayer’s haematoxylin (Lillie’s modification; DAKO), washed in dH2O, and incubated for 30 s in 0.05% ammonium solution (PBS) followed by washing in dH2O. Before mounting the coverslips with VectaMount aqueous mounting medium (VectorLab, H-5501-60), the slides were dehydrated by washing for 1 min in 75% ethanol, 5 min in 100% ethanol and 5 min in Histoclear. Slide images were acquired using a ×20 bright-field objective on a Zeiss AxioScan Z.1 slide scanner, and analysis was performed on fields using QuPath version 0.2.0-m9 using an in-built positive cell detection tool to segment haematoxylin-stained nuclei and quantify the mean intensity of DAB.
Histologic analysis of the mouse fibrosis experimentLeft lung tissue was fixed in a 10% neutral buffered formalin solution for 24 h and subsequently transferred into tissue cassettes and placed into PBS for a minimum of 24 h. The tissues were then shipped to the Institute for Research in Biomedicine (IRB) Histopathology Facility for paraffin embedding, sectioning and Masson’s trichrome and haematoxilin and eosin staining. Samples were examined first in a blinded fashion and in a second round in an unblinded fashion. Semiquantitative histological scoring of fibrosis was scored at ×20–40 using the following scale: 1, ×1; 2, ×2; 3, ×3 increase in the thickening of alveolar walls; 4, >×3 thickening of alveolar walls and focal areas of single fibrotic masses. If there was difficulty in deciding between two scores, the intervening number was given.
Hydroxyproline assaySuperior and middle lung lobes were surgically dissected, weighed and placed into 1.5-ml sterile tubes and flash-frozen until all the samples were collected. Homogenates of the lung samples were made by grinding the frozen samples with liquid nitrogen using a mortar and pestle. On the day of the assay, tissues were thawed, and 1 ml of distilled water was added to the tissues. Tissues were homogenized using a micro-sample homogenizer (Precellys), then 200 µl of 12 N hydrochloride was added to 200 µl of homogenized tissues. The samples were placed into a preheated oven set to 120 °C and incubated overnight. The next morning, samples were cooled and vortexed. Biochemical quantification of hydroxyproline was performed using a hydroxyproline assay kit (Amsbio).
Senescence-associated β-galactosidase assayCells grown in six-well plates were fixed with a solution of 0.5% glutaraldehyde (wt/vol, PBS; Sigma) for 10 min and washed twice in a solution of 1 mM MgCl2/PBS (pH 6.0). For staining, the plates were incubated with X-gal staining solution for 18 h at 37 °C. Images were acquired by bright-field microscopy using an inverted microscope (Olympus CKX41) with an attached digital camera (Olympus DP20). Cells were counted using ImageJ software to determine the percentage of positive cells.
Liver samples frozen in OCT were cryosectioned (15 μM), and the frozen sections were fixed in ice-cold 0.5% glutaraldehyde (wt/vol, PBS) for 15 min and washed 1 mM MgCl2/PBS (pH 6.0) for 5 min. The β-galactosidase activity was stained for with X-gal staining solution (1 mg ml−1 X-gal, Thermo Scientific, 5 mM K3(Fe(CN)6), 5 mM K4(Fe(CN)6)) diluted in 1 mM MgCl2/PBS (pH 6.0) for 18 h at 37 °C. Slides were dehydrated and coverslips mounted before being imaged using ×20 bright-field objective on a Zeiss AxioScan Z.1 slide scanner. ImageJ was used to quantify staining by measuring the SA-β-Gal-stained area as a percentage of the total tissue area excluding luminal spaces.
Sirius Red stainingSirius Red staining was carried out for collagen I/III fibre-containing connective tissue on paraffin-embedded sections using a Picrosirius Red stain kit (Abcam, ab150681). Before staining, sections were deparaffinized in Histoclear and graded ethanol washes as already described (IHC staining section), then hydrated in distilled water. Sections were then incubated with Picrosirius Red solution for 60 min at r.t. and then rinsed twice with 0.5% glacial acetic acid solution (in dH2O). Excess water was then removed by shaking the slides and then rinsing in 100% ethanol. Sections were then dehydrated by two washes of 100% ethanol for 2 min each and two washes in Histoclear for 2 min each. Coverslips were mounted and slides were imaged on a Zeiss AxioScan Z.1 system. Staining was quantified by thresholding the collagen-stained area for detection of fibres (red) and measuring this area relative to the total tissue area.
Blood chemistry and immune cell composition analysisFor analysis of immune cell composition in whole blood, tail-vein blood was collected two days after the last treatment. Whole blood was diluted in saline to a volume of 200 μl and run on a Sysmex XE2100 automated cell counter. Blood glucose levels were determined by collecting whole blood from the tail vein into heparinized tubes (Abraxis), then 120–140 μl of whole blood was loaded onto a comprehensive diagnostic profile reagent rotor (Abraxis) or Mammalian Liver Profile reagent rotor and run on a VetScan VS2 Chemistry Analyzer (Abraxis, 500-7123).
Oil Red O stainingStaining for lipids was carried out on liver tissue in OCT that was snap-frozen in liquid N2 and cryosectioned (15 µm). Sections were equilibrated to r.t. for 10 min and then stained with 0.5% Oil Red O solution (wt/vol, in isopropanol; Sigma, O1391) for 5 min, rinsed in tap water and counterstained with Mayer’s haematoxylin for 30 s. Sections were then rinsed again in tap water for 30 min and coverslips mounted. Images were acquired on a Zeiss AxioScan Z.1 system and ImageJ quantification of the Oil Red stain area was carried out relative to the background tissue area.
Ex vivo culture of mouse pituitariesNeoplastic pituitaries from 18.5dpc Hesx1Cre/+;Ctnnb1lox(ex3)/+ embryos were dissected and placed on top of 5 μM Nuclepore membranes (VWR) in 24-well plates containing 500 μl of medium (DMEM-F12 (Gibco), 1% pen/strep (Sigma) and 1% FBS (Thermo Fisher Scientific)) supplemented with either IMP1088 or vehicle (DMSO). The media were changed every 24 h, and the pituitaries were processed for analysis after 72 h. IF staining was performed as described in ref. 53. The proportion of β-catenin-accumulating cells was calculated as an index out of the total DAPI-stained nuclei. Over 120,000 DAPI nuclei were counted from 15 to 22 histological sections per sample, in a total of eight neoplastic pituitaries. The proportions of CC3 and synaptophysin-positive cells were calculated as an index out of the total tissue area, from 6 to 12 histological sections per sample.
ImmunoblottingCells were collected for protein extraction by first washing twice with ice-cold PBS, scraping, then centrifugation performed at 180g for 5 min at 4 °C. Cell pellets were then resuspended in RIPA lysis buffer (Thermo Scientific, 89900) supplemented with one tablet of PhosSTOP (Roche) and one tablet of cOmplete, Mini, EDTA-free Protease inhibitor (Roche). Lysis was performed on ice for 30 min with periodic vortexing. The lysis samples were centrifuged at 14,500g for 20 min at 4 °C and protein-containing supernatant was transferred to a fresh tube. RIPA lysed samples quantification was then performed using a Pierce BCA assay (Thermo Scientific) and equal amounts of sample was resuspended in required volumes of 4× Laemmli sample buffer (Bio-Rad, 1610747) and boiled at 95 °C for 10 min. To immunoblot the proteins, samples were separated by size on pre-cast polyacrylamide gradient gels (Bio-Rad, 4561084) and transferred onto 0.2-µm nitrocellulose membranes (Bio-Rad). Efficient transfer and correct gel loading were verified by Ponceau S staining before 1 h blocking of membranes with 5% milk (wt/vol) diluted in TBS supplemented with 0.1% Tween-20 (vol/vol; TBST). Primary antibodies were diluted in 5% milk (wt/vol, TBST) and incubated with membranes overnight at 4 °C. This was then followed by three washes with TBST followed by 1 h incubation with HRP-conjugated secondary antibody. Secondary antibody binding was visualized using Amersham ECL Prime western blotting detection reagent (Cytiva) and imaged on an Amersham Imager 680 blot and gel imager (Cytiva).
RNA extractionTotal RNA from the tissues was extracted in a bulk way by bead disruption in 800 µl of TRIzol reagent (Invitrogen) using a TissueLyser system (Qiagen) followed by further homogenization using a QIAshredder kit (Qiagen), according to the manufacturer’s instructions. The homogenized tissue in TRIzol was then mixed with 160 µl of chloroform (Sigma) and vortexed for 15 s, then centrifuged at 14,500g at 4 °C for 30–45 min. The top aqueous phase containing RNA was then column-purified using an RNAeasy Mini Kit (Qiagen) and subjected to DNase treatment according to the manufacturer’s instructions. RNA concentration was determined using a NanoDrop ND-1000 UV–vis spectrophotometer at a wavelength of 260 nm.
For extraction of total RNA from cells, six-well plates were scraped in 800 µl of TRIzol reagent (Invitrogen), mixed with 160 µl of chloroform (Sigma), vortexed and centrifuged as stated above. The aqueous phase was then transferred to a new tube and processed from step 2 onwards of the manufacturer’s instructions for the RNAeasy Mini Kit (Qiagen).
cDNA synthesis and quantitative RT–PCRTo generate cDNA, total RNA was diluted in nuclease-free water to the same concentration across samples of the same experiment, and 1–5 µg was amplified using a SuperScript II reverse transcriptase kit (Invitrogen) combined with 1 µl of random hexamer primers (50 ng µl−1, Invitrogen), 1 µl dNTP mix (10 mM, Bioline) and made up to a final volume of 11 µl in nuclease-free water. The mixture was then added to a thermocycler for one cycle of 10 min at 25 °C, 50 min at 42 °C and 15 min at 70 °C. cDNA samples were then diluted at 10 ng µl−1 based on input RNA concentration.
mRNA expression analysis was carried out using real-time quantitative PCR (RT–qPCR) by way of amplification of cDNA using SYBR Green PCR Master Mix (Applied Biosystems) run on a CFX96 Real-Time PCR Detection system (Bio-Rad). RT–qPCR primers were selected from PrimerBank69 spanning exon–exon junctions. The relative gene expression in human cell lines was determined using the ΔΔCt method by measuring the RT–qPCR signal relative to the signal of the housekeeping gene RPS14 and normalization to control samples. For mouse mRNA expression, the ΔΔCt method was again used, but the signal was measured relative to GAPDH.
For the mouse fibrosis experiments, tissues were homogenized in TRIzol and the cDNA was synthesized using SuperScript III reverse transcriptase (Thermo Fisher). Real-time PCR was performed using the PowerUp SYBR Green Master Mix (Applied Biosystems). Gene expression analysis was performed using the indicated primers. The results were then normalized using the housekeeping gene Gapdh, Actin b or Hprt. The primer pairs used are presented in Supplementary Table 2.
RNA-seq and GSEAThe total RNA extracted and purified from tissues or cell extraction was analysed on a 2100 Bioanalyzer (Agilent) using an RNA 6000 Nano Kit (Agilent) to verify the RNA purity and integrity before library preparation. RNA from tissue samples with an RNA integrity number (RIN) corresponding to a ratio of the 18S-to-28S rRNA peaks on the bioanalyser trace of less than 3 were not submitted for library processing. Library preparation to generate cDNA was performed by the MRC–LMS genomics core facility with 200 ng of starting RNA using the NEBNext Poly(A) mRNA magnetic isolation kit (NEB, E7490) to isolate mRNA from the total RNA sample. Purified samples were then processed using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB, E7760). Libraries were then assessed on a 2100 bioanalyzer and concentration was determined using a Qubit fluorometer and the Qubit dsDNA HS assay kit (Thermo Scientific). Indexed libraries were then run on two lanes of a NextSeq 2000 sequencer (Illumina), with >10 million single-end 75-bp reads being generated per sample. Human RNA-seq reads were assessed for quality using FASTQC and then aligned to human genome hg19 by Tophat (v. 2.0.11) using ‘-library-type- fr-firststrand’ parameters along with gene annotation from Ensembl (v.67). GSEA was carried out on the differential expression of vehicle and drug-treated aged tissues using Wald statistics parameters in DESeq2 and all curated gene sets in MSigDB.
Live-cell microscopyTo analyse the live-cell induction of apoptosis, cells were incubated with IncuCyte caspase-3/7 reagent (1:500, Essen Bioscience) following reverse transfection with senolytic siRNAs or drug treatment. Four images per well of a 96-well plate were collected every 2 h for 3–4 days using a ×10 objective on an IncuCyte microscope, and fluorescence images were analysed with IncuCyte Zoom software (Essen Bioscience).
Druggable genome siRNA screening and siRNA transfectionDruggable genome siRNA libraries were purchased from Qiagen (Human Druggable Genome siRNA Set V4.1, 2 siRNA per gene) and Dharmacon (siGenome human druggable genome, four siRNA per gene). Individual siRNAs were purchased from the siGenome reagent family of Dharmacon (Horizon Discovery) and came lyophilized in tube format or coated onto 96-well plates. Before transfection, plates containing 0.1 nM of lyophilized siRNA were resuspended in 100 µl of nuclease-free water and 3.6 µl of siRNA aliquoted into daughter plates. For large-scale libraries, daughter plates were aliquoted using a laboratory automation workstation (Biomek NXP, Beckman Coulter). Transfection mix containing 0.2 µl of DharmaFECT 1 with 17.4 µl of DMEM only or 0.4 µl DharmaFECT 1 with 17.2 µl of DMEM was added to daughter siRNA plates for IMR90 ER:RAS or IMR90 experiments, respectively. To reverse-transfect cells, 100-µl suspensions of proliferating or senescent cells in medium with DMEM supplemented with 10% FBS only were added to plates with combined transfection mix and siRNA (final siRNA concentration 30 nM). After 18 h, when cells had been allowed to adhere, the medium was replaced with DMEM supplemented with 0.5% (wt/vol) FBS and 1% antibiotic–antimycotic solution. Plates were then fixed in 4% PFA (wt/vol) 72 h after a medium change, to then be processed for quantitative IF. For analysis of mRNA, the protocol was scaled to a six-well-plate format and cells were collected by the addition of TRIzol RNA isolation reagent (Invitrogen) to the well followed by scraping and collection. Information about the siRNAs used in this study is provided in Supplementary Table 3.
B-score normalization analysisTo analyse the siRNA screen, cell counts were normalized by B-score using the R package CellHTS2 (https://doi.org/10.18129/B9.bioc.cellHTS2)70. Cell count normalization was performed using the plate-averaging method and on separate batches for control and senescent cells, in addition to a separate normalization performed for each batch of plate transfections.
Enzyme-linked immunosorbent assayFor the detection of secreted factors in conditioned media of IMR90 ER:RAS cells, 100 µl of medium (DMEM supplemented with 0.5% (wt/vol) FBS and 1% antibiotic–antimycotic solution) incubated with cells and inhibitors for 48–72 h was collected and filtered using a 0.2-µm cellulose acetate membrane (Gilson). Filtered samples were then subject to an enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s instructions (R&D: IL-6, DY206; IL-8, DY208; VEGF, DY293B; CXCL1, DY275; G-CSF, DY214; GM-CSF, DY215; CCL2, DY279; CCL20, DY360; LIF, DY7734). Cell numbers were calculated using high-throughput microscopy and used to normalize the levels of secreted factors.
Proteostat assayRelative levels of protein aggregates were measured using the PROTEOSTAT protein aggregation assay (ENZ-51023) according to the manufacturer’s instructions. Briefly, cells plated in a 96-well format and treated with the drug were incubated with PROTEOSTAT detection reagent for 15 min at r.t. and read on a FLUOstar Omega plate reader at 550 nm (ex.) and 600 nm (em.). The background was subtracted and intensity values normalized to cell counts from fixed DAPI-stained plates using high-throughput microscopy.
Visualization of IpaJ effects on N-myristoylation with YnMyr and immunoblot analysisIn triplicate for each condition, IMR90 cells (controls, and cells transduced with IpaJ WT and IpaJ C64A constructs) were seeded in six-well plates and grown to 70–80% confluence. IMR90 control cells were incubated for 1 h with DMSO or 100 nM IMP1088. All conditions, including IpaJ variant-expressing cells, were thereafter metabolically labelled with 20 μM YnMyr for 18 h. The cells were then washed with PBS, collected by trypsinization, and the cell pellets were stored at −80 °C until further analysis. The cell pellets were lysed and the YnMyr-labelled proteins were functionalized with fluorescent capture reagent, then resolved by fluorescence scanning after separation on 15% (wt/vol) SDS–PAGE gels as previously described45,50. Immunoblotting was performed on ARF1, ARL1, PPM1β and TUBA, then read out on a Li-Cor Odyssey CLx system using IRDye 800CW-functionalized secondary antibodies. Fluorescence intensities were quantified by ImageJ and normalized to the TUBA loading control.
Statistics and reproducibilityStatistical analyses were performed and plotted using GraphPad Prism 9 software. Details of the test used are given in the corresponding figure legends and the source data. Statistical analysis was performed using either an unpaired two-tailed t-test with Holm–Sidak multiple comparison correction or with ordinary one- or two-way ANOVA with Dunnett’s or Tukey’s multiple comparison correction. Tumour growth curves were analysed using Repeated Measure (RM) two-way ANOVA with Greenhouse–Geisser correction and Dunnett’s correction. P values and adjusted P values are shown for values lower than P = 0.1. P values and adjusted P values for other comparisons and per experiment statistical test details are available in the source data.
No statistical method was used to predetermine sample size. For the bleomycin-induced fibrosis experiment, two mice in the vehicle-treated group died before the end of the experiment and were not included in any analysis. For the WD experiment, a mouse of the WD + vehicle group died before the end of the experiment and was not included in any analysis. RNA isolation from one mouse liver of the WD + NMTi group failed and it could not be included in subsequent RT–qPCR analysis. Three sections from Fig. 8h were excluded due to poor section processing. No further data were excluded from analyses. For in vivo studies, mice were randomized to treatment groups. Cell-culture experiments were not randomized. Histologic analysis of Masson’s trichrome and haematoxilin & eosin (H&E) staining for the mouse fibrosis experiment was examined first in a blinded fashion and in a second round in an unblinded fashion. Tumour measurements were taken blindly and independently by the researcher or the animal technicians in the mouse facility. For the HDTVI and WD experiments, staining and analysis were performed in a blinded fashion. Investigators were not blinded during the other experiments.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this Article.
Comments (0)