Cell culture and cell lines
All cells were cultured at 37°C with 5% CO2 in DMEM (Gibco, 10569-010) supplemented with 10% fetal bovine serum (Gibco, 10270106). Wild-type and ΔEMC6 HEK293 cells have been previously described14. The ΔTMCO1 HEK293 cell line was obtained from R. Keenan20. ΔEMC6ΔTMCO1 double-knockout cells were generated by knocking out TMCO1 in ΔEMC6 cells. Ribonucleoprotein complexes were formed between Alt-R S.p. Cas9-GFP V3 (IDT, 10008100) and Alt-R CRISPR-Cas9 sgRNA (5′-ACTTGTCTGTCCTGTAAACC-3′; IDT) following the manufacturer’s recommendations. Ribonucleoprotein complexes were transfected into ΔEMC6 cells using Lipofectamine RNAiMAX (Invitrogen, 13778150) according to the manufacturer’s protocol; 48 h later, green fluorescent protein (GFP)-positive cells were sorted into single colonies and expanded. Knockout cells were screened by immunoblotting. Flp-In T-REx 293 cells stably expressing wild-type EMC3-FLAG (NP_060917.1) or EMC3-FLAG variants (Mcyt-1-S: M101, 106, 110, 111S; R31A; F148L; R13E) were generated by integrating each construct into the FRT site and selecting for Flp-mediated recombination through 100 µg ml–1 hygromycin B for 2 weeks6. The tetracycline-inducible 293 cell line expressing the human GABAA receptor has been previously described29. In this cell line, the α1 subunit (NP_001178048.1) in the pcDNA4-TO-Zeocin backbone is FLAG-tagged after its 27 amino acid signal sequence; the β3 subunit (NP_068712.1) in a pcDNA3.1-TO-Hygromycin backbone is untagged; the γ2L subunit (NP_944494.1) in a pACMV-TO-blasticidin backbone is 1D4-tagged (TETSQVAPA) at the C terminus after a (GGS)3GK linker. Tetracycline-inducible 293 cell lines expressing GFP-P2A-RFP-SQS (NP_004453.3, aa378–410) and GFP-P2A-RFP-ASGR1 (NP_001662.1) reporters in the pcDNA5-FRT-TO backbone were generated by stably integrating reporter plasmids into the FRT site and have been previously described4.
Recombinant DNA reagents
Plasmids or gBlocks (IDT) used for in vitro transcription and translation assays contained an SP6 promoter and coding sequences. All plasmids were verified by sequencing. Wild-type GABRA1 (NP_001345964.1) contains two mutations (V436M and L448M) to facilitate the detection of the C-terminal domain by autoradiography without changing the TMD length, hydrophobicity or C-tail charge. GABRA1-glyc was generated by adding an opsin tag (MNGTEGPNFYVPFSNKTVD)66 to the C terminus of wild-type GABRA1. SQS-glyc (NP_004453.3, aa378–410) has been previously described14. The 23L-GABRA1 contains an N-terminal 9×His tag, a glycosylation sequence, a soluble tail from β1-adrenergic receptor (NP_001290104.1, residues 29–44), 23 leucine codons, a soluble cytosolic loop (GGSG-mEGFP(1–92)), TMD4 and flanking regions of GABRA1 (NP_001345964.1, aa 407–455) and an opsin tag. 23L-SQS replaces TMD4 and flanking sequences of 23L-GABRA1 with the TMD and flanking regions of SQS (NP_004453.3, aa378–410). The following 23L-SQS variants, used in Figs. 3 and 4, were made by site-directed mutagenesis: P202C (cysteine in C-tail for EMC crosslinking); P202Amber (for incorporation of a photoreactive amino acid into the C-tail); S168C (cysteine in TMD); S185E (−3); E189R (0); E189R,D190R (+2); S185R,E189R,D190R (+3); T183L (1L); T182L,T183L (2L); Q179L,T182L,T183L (3L); S177L,Q179L,T182L,T183L (4L); and S168L,S177L,Q179L,T182L,T183L (5L). Extensions of the C-tail length to a total length of 25, 35, 45, 55, 65 and 225 amino acids were generated by inserting part of the coding sequence for mCherry. The Rho(1–3) domain that precedes the cytosolic loop in the GABRA1 and SQS constructs shown in Fig. 5 contains the following: the prolactin signal sequence (NP_776378, aa1–33), a Twin-Strep-tag (SAWSHPQFEKGGGSGGGSGGSAWSHPQFEK), a linker (AGGSAGSGGGSAGGSA), the VHP domain (NP_990773, aa792–826), a glycosylation site, a linker (GGGSAGGGSA) and rhodopsin (NP_001014890, aa32–152). For the reporter constructs shown in Fig. 7b, TMD4 and flanking regions of 23L-GABRA1 were replaced by the following sequences: CNIH2 (NP_872359, aa126–160); VATL (NP_001685, aa119–155); S38A1 (NP_109599, aa440–487); ACHA4 (NP_000735, aa588–627); 5HT3B (NP_006019, aa402–441); and GLRB (NP_001159532, aa463–497). SOAT1 (NP_003092.4) was appended at the C terminus with the opsin glycosylation tag. The N-terminal deletion of SOAT1 removed amino acids 2–125. YIPF1 (NP_061855.1) was obtained from R. Keenan.
Small interfering RNA knockdown and flow cytometry
For monitoring the surface expression of GABAA receptors, either negative control small interfering RNA (siRNA) (Invitrogen, 4390843) or EMC4 siRNA (Ambion, s27733) was transfected into the 293 cell line expressing the human GABAA receptor using Lipofectamine RNAiMAX according to the manufacturer’s protocol. A final concentration of 10 nM of siRNA was used and the total knockdown time was 66 h. At 60 h, doxycycline was added to a final concentration of 0.1 µg ml–1 to induce expression of the GABAA receptor for 6 h. Cells were then collected and subjected to surface labeling. Pelleted cells were resuspended with 100 µl of cold PBS, supplemented with 1 µl of phycoerythrin-labeled FLAG antibody (BioLegend, 637310) and incubated in the dark at 4°C for 1 h. Cells were then washed with cold PBS, passed through a 70 µm filter and then analyzed on a BD LSR II flow cytometer for appropriate fluorescent channels. A total of 30,000 events were analyzed, and phycoerythrin fluorescence, reflective of GABAA receptor surface levels, was plotted as a histogram using FlowJo. To analyze the stability of ASGR1 or SQS, 293 cell lines stably expressing GFP-P2A-RFP-ASGR1 or GFP-P2A-RFP-SQS were used as previously described50. The P2A sequence in these constructs causes ribosome skipping, resulting in the translation of equimolar amounts of GFP and the RFP-tagged protein. Therefore, a steady-state RFP:GFP ratio reflects the stability of the RFP-tagged protein. Failure in biogenesis will lead to degradation by cellular quality control pathways and a decreased RFP:GFP ratio. Knockdown was performed as for GABAA receptor cells, with the GFP and RFP fluorescence monitored by flow cytometry on 30,000 cells. The RFP:GFP ratio was plotted as a histogram using FlowJo.
Preparation of SPCs
Cells at 95–100% confluency were trypsinized and collected by centrifugation at 4 °C, washed once with ice-cold 1×PBS and resuspended in 1×RNC buffer (50 mM HEPES, pH 7.4, 100 mM KOAc, 5 mM Mg(OAc)2) containing 0.01% purified digitonin67. SPCs were pelleted and washed once with 1×RNC. To digest endogenous mRNAs, SPCs were resuspended in 100 µl of 1×RNC containing 1 mM CaCl2 and 150 units per ml micrococcal nuclease (Roche, 10107921001). Nuclease digestion was performed for 10 min at room temperature (20 °C) and was terminated by adding a final concentration of 2 mM EGTA. Nuclease-digested SPCs were pelleted, washed once with 1×RNC buffer, resuspended in 0.5×RNC buffer to 6,000–10,000 cells per ml and used immediately in translocation assays.
Preparation of endoplasmic-reticulum-enriched membranes
Approximately 80% of confluence cells were collected by trypsinization. Cells were pelleted, washed once with cold 1×PBS and flash-frozen in liquid nitrogen. Thawed cell pellets were mixed with 5 volumes of 20 mM HEPES, pH 7.4; 5 mM KCl; 1.5 mM MgCl2; 2 mM dithiothreitol (DTT); and protease inhibitor (Roche, 10106399001) and incubated on ice for 15 min. Cells were lysed on ice by 35 strokes of dounce homogenization (DWK Life Sciences, 357542). Cell lysates were adjusted to 20 mM HEPES, pH 7.4; 210 mM mannitol; 70 mM sucrose; 0.5 mM EDTA; 2 mM DTT; and protease inhibitor. Cell debris and nuclei were cleared by centrifugation at 4 °C for 10 min at 700×g. Membranes were then pelleted by centrifugation at 4°C for 10 min at 8,500×g, washed once and resuspended in a buffer containing 20 mM HEPES, pH 7.4; 210 mM mannitol; 70 mM sucrose; 0.5 mM EDTA; 2 mM DTT; and protease inhibitor to give an OD280 of 20. Different amounts of resuspended membranes were used to assay the levels of key translocation components by blotting.
In vitro transcription and translation
Transcription reactions with SP6 polymerase were performed at 37°C for 1 h and contained the following components: DNA that encodes regions of interest for translation reactions (PCR-amplified and purified by Qiagen PCR purification kit, 10 ng µl–1); HEPES, pH 7.4 (40 mM); spermidine (2 mM; Sigma, S0266); RNA cap structure analog (0.33 mM; NEB, S1404L); reduced glutathione (10 mM); MgCl2 (6 mM); NTPs (0.5 mM each for ATP; Roche, 10519979001), CTP (Sigma, C1506) and UTP (Sigma, U6875), 0.1 mM for GTP (Roche, 10106399001); SP6 RNA polymerase (0.4 U µl–1; NEB, M0207L); and RNase inhibitor (0.8 U µl–1; Promega, N2515).
Translation reactions were performed at 32°C for 30 min and contained the following components: micrococcal nuclease-digested rabbit reticulocyte lysates (Green Hectares) (34% of the total volume); transcription reaction from the previous step (5% volume); SPCs (10% volume); ATP and GTP (1 mM each); an ATP regeneration system (creatine phosphate (12 mM; Roche, 10621714001); creatine kinase (0.04 mg ml–1; Roche, 10127566001)); spermidine (0.3 mM); HEPES, pH 7.4 (20 mM); KOAc (50 mM); Mg(OAC)2 (2 mM); reduced glutathione (1 mM); tRNAs purified from pig liver (0.05 mg ml–1); 19 of the 20 amino acid except for methionine (40 µM each; Promega, L9961); and 35S-methionine (0.5 µCi µl–1; PerkinElmer, NEG709A001MC).
For incorporating the photoreactive amino acid p-benzoyl-l-phenylalanine (Bpa) through amber suppression, the following components are included in the translation reaction68: suppressor Bacillus stearothermophilus tRNACUATyr (5 µM); E. coli Bpa tRNA synthetase (0.25 µM); and Bpa (100 µM). These components were pre-mixed into a 10× solution (in 50 mM HEPES, pH 7.4, 100 mM KOAc, 1 mM Mg(OAC)2) and were pre-incubated at 32°C for 15 min before adding to the translation reaction. Where indicated, the Sec61 lateral gate inhibitor ApraA (obtained from V. Paavilainen and K. McPhail) was included in the translation reaction at 2 µM.
Protease protection assays
The 60 µl in vitro translation reactions were chilled on ice, and the SPCs were pelleted (20,000×g for 2 min), washed once with 1×RNC buffer (50 mM HEPES, pH 7.4, 100 mM KOAc, 5 mM Mg(OAc)2) and resuspended in 30 µl 0.5×RNC buffer. Samples lacking SPCs were used directly without pelleting. Samples were divided into two aliquots; one aliquot (two-thirds of the total volume) was adjusted to 0.5 mg ml–1 proteinase K and incubated on ice for 50 min. Proteinase K was quenched by adding 250 mM of PMSF for 2 min, then transferring the entire reaction to a tenfold excess volume of 1% SDS, 100 mM Tris-HCl, pH 8.0 pre-heated to 100°C and heated for 10 min. The samples were either analyzed directly or subjected to immunoprecipitation as indicated in the figure legends.
Site-specific crosslinking
The 120 µl in vitro translation reactions were used for bismaleimidohexane (BMH) crosslinking experiments. All steps following the translation reaction were at 4°C until the reaction was denatured in SDS. SPCs were pelleted and resuspended in 60 µl 0.5×RNC buffer. One aliquot was removed as the no-crosslinking control and the remainder of the sample was adjusted to 250 µM BMH (Thermo Scientific, 22330) and incubated on ice for 10 min. The crosslinking reaction was quenched by adjusting the final concentration of DTT to 25 mM. After denaturation in 1% SDS, 100 mM Tris-HCl, pH 8.0, the samples were either analyzed directly or processed further for immunoprecipitation or deglycosylation. SMPH (Succinimidyl 6-((beta-maleimidopropionamido)hexanoate)) and UV crosslinking experiments were performed similarly to the BMH crosslinking experiments, with the following differences: SMPH (Thermo Scientific, 22363) crosslinking (200 µl total reaction) was at 200 µM final concentration for 30 min, and quenched with 50 mM Tris-HCl pH 7.4 and 5 mM DTT; UV crosslinking (100 µl total reaction) was on ice with UV irradiation by a UVP Blak-Ray B-100AP high-intensity lamp with the bulb positioned ~10 cm above the samples.
Immunoprecipitation and PNGase F treatment
Denaturing immunoprecipitation after crosslinking, proteinase K digestion and glycanase digestion have been previously described6. SDS-denatured samples were diluted tenfold in ice-cold immunoprecipitation buffer (1×PBS, 250 mM NaCl, 0.5% TX-100, 10 mM imidazole) and mixed with 2.5 µl of anti-FLAG resin (Millipore, A2220), 2.5 µl of Monoclonal Anti-HA resin (Millipore, A2095), or 5 µl of protein A resin (Repligen, CA-HF-0100) along with the appropriate antibody. A total of 1.25 µg of GABRA1 antibody (Invitrogen, PA5-79291) was used per immunoprecipitation. The mixture was rotated end-over-end for 1.5 h (for FLAG or HA) or 3 h (for GABRA1 immunoprecipitation) at 4°C. Beads were washed twice with cold immunoprecipitation buffer and eluted by boiling in 10 µl of 2.5× SDS–PAGE sample buffer for 10 min. For deglycosylation experiments, crosslinked samples (Fig. 3) or total translation reactions (Extended Data Fig. 4) were split into two halves after denaturation with 0.5% SDS and 50 mM Tris-HCl, pH 8. One half was untreated and the other was adjusted to 1% NP-40, 1× GlycoBuffer 2 and 25 U ml–1 of PNGase F (NEB, P0704S) and digested at 32°C for 30 min. Both halves were subjected to immunoprecipitation as described above (Fig. 3) or analyzed directly (Extended Data Fig. 4).
Bioinformatic analysis of membrane proteins
All proteins containing TMDs were retrieved from the UniProt database69. The UniProt annotations were used to define the start and end of the TMD helices. Proteins containing a single TMD or multipass membrane proteins localized to mitochondria were manually removed from this set. The AlphaFold2 (ref. 51) predicted structure, available from the UniProt database for each of the remaining 1,784 multipass membrane proteins, was inspected manually to annotate the number of TMD helices and the overall charge of each TMD-flanking side of the structure. The overall basic flank was designated cytosolic as per the positive-inside rule52. Then the C-terminal TMD was identified and assigned the appropriate orientation, hydrophobicity (as calculated using the ∆Gapp predictor65) and flanking C-tail length. C-tails facing the cytosol were designated ‘Ccyt’ and those facing the opposite orientation were designated ‘Cexo’. The curated list is provided in Supplementary Table 1; information from this table was used to generate the plots in Fig. 6.
SDS–PAGE and blotting
Cell lysates or endoplasmic-reticulum-enriched membranes were analyzed by SDS–PAGE on 12% Tris-Tricine gels. SDS–PAGE gels were transferred to a nitrocellulose membrane (Biorad, 1620112) and blotting was performed with standard procedures using 5% non-fat dried milk as the blocking agent. The following antibodies and dilutions were used for blotting: CCDC47 (Bethyl Laboratories, A305-100A; 1:5,000); EMC3 (Invitrogen, 711771; 1:5,000); EMC6 (Abcam, ab84902; 1:1,000); Calnexin (Enzo, ADI-SPA-865; 1:5,000); Sec61α (ref. 70; 1:5,000); TMCO1 (Invitrogen, PA5-43350; 1:500); Sec61β (ref. 71; 1:5,000); CAML (Cell Signaling Technology, 13913S; 1:1,000); FLAG M2-HRP (Sigma, A8592; 1:5,000); EMC4 (Abcam, ab123719; 1:2,000); and β-Actin-HRP (Sigma, A3854; 1:10,000).
Quantification of C-tail translocation
Quantification was performed on raw phosphorimager files using Fiji. The pixel intensity and area of each band were measured, from which the background intensity was subtracted. For the C-terminal TMD reporters, C-tail translocation was calculated by dividing the value for the C-tail translocated band (typically the 2×-glycosylated product) by the sum of total membrane inserted bands (typically the 1×-glycosylated and 2×-glycosylated bands). In the case of SQS-glyc (Fig. 1d,e), SOAT1 (Fig. 7c) and YIPF1 (Fig. 7d), per cent C-tail translocation is calculated by dividing the intensity of the glycosylated band by the sum of glycosylated and non-glycosylated bands.
Statistics and reproducibility
This study does not contain any statistical analysis. All data presented in this paper have been reproduced in independent experiments. The number of independent experiments are indicated in parentheses for the following main and extended data figure panels: Fig. 2a (2); Fig. 2b (2); Fig. 2c (2); Fig. 2d (3); Fig. 3a (3); Fig. 3b (3); Fig. 3c (2); Fig. 3d (3); Fig. 4a (2); Fig. 4b (2); Fig. 4c (2); Fig. 5 (2); Fig. 7b (2); Fig. 7c (4 for SOAT1, 2 for SOAT1ΔN); Fig. 7d (6); Extended Data Fig. 1 (2); Extended Data Fig. 2 (2); Extended Data Fig. 3 (2); Extended Data Fig. 4 (2).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.


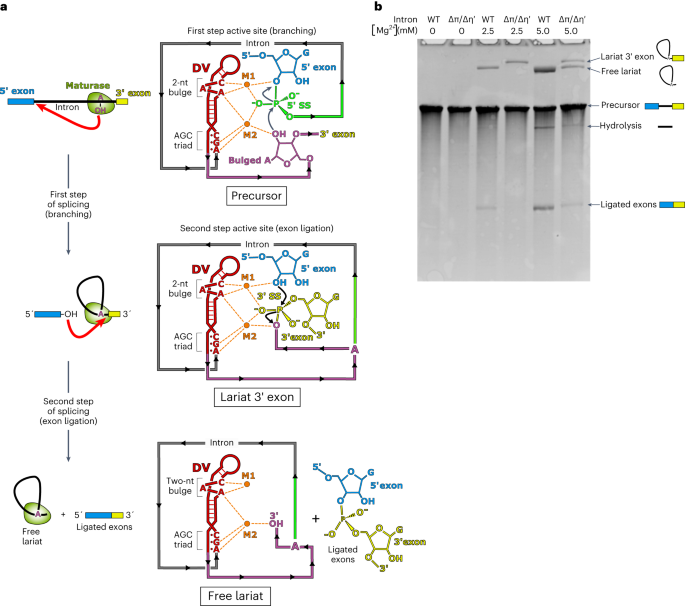
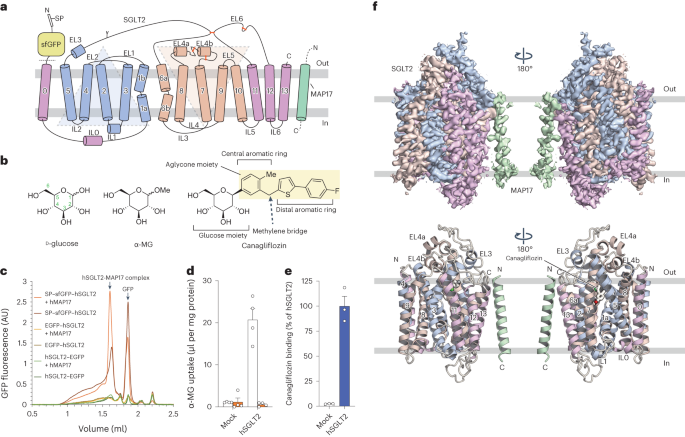
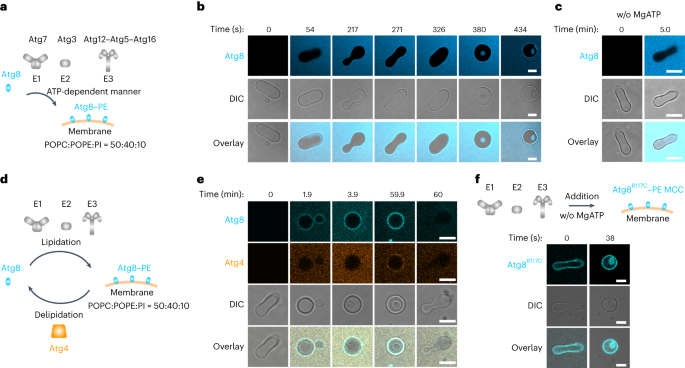
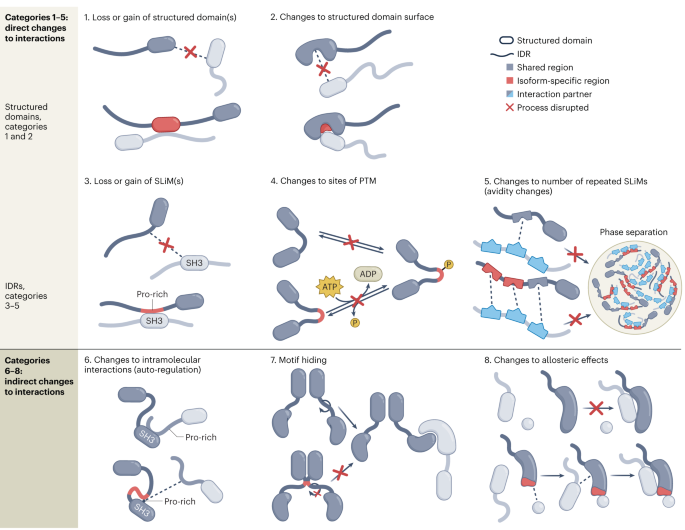
Comments (0)