
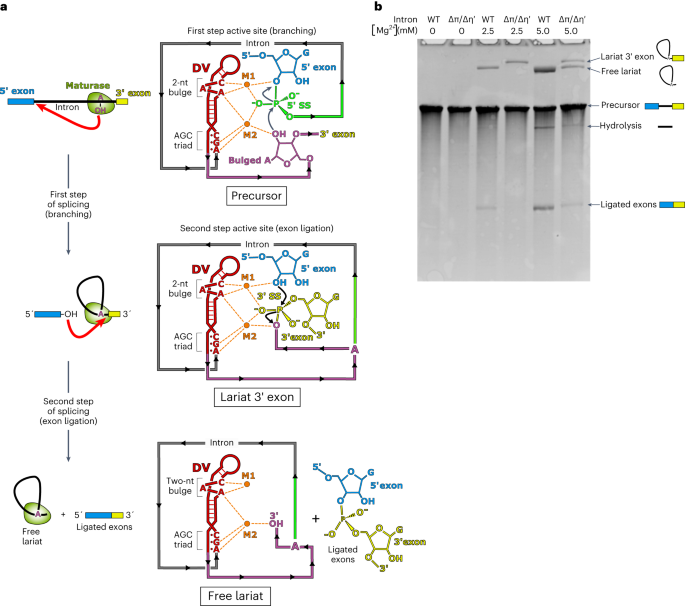
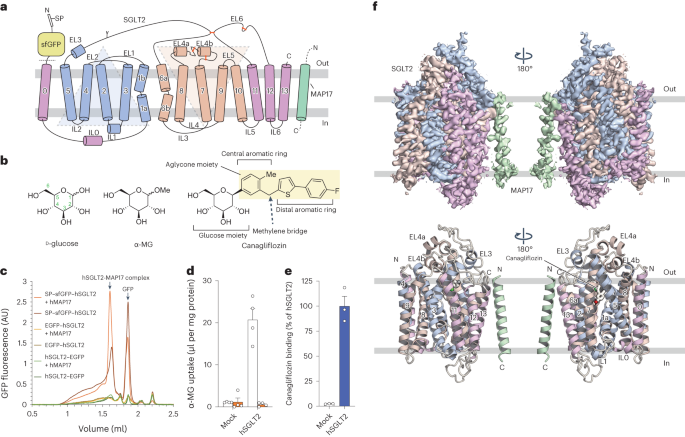
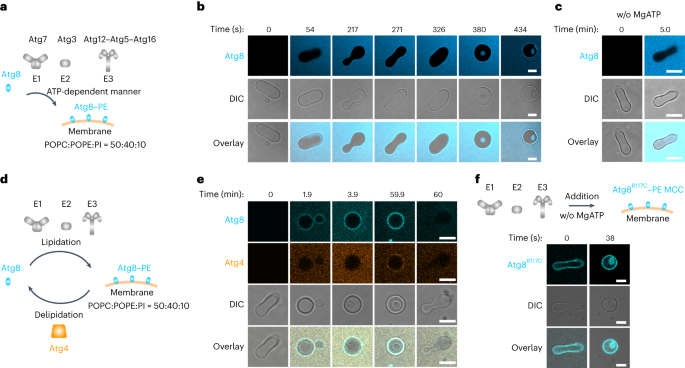
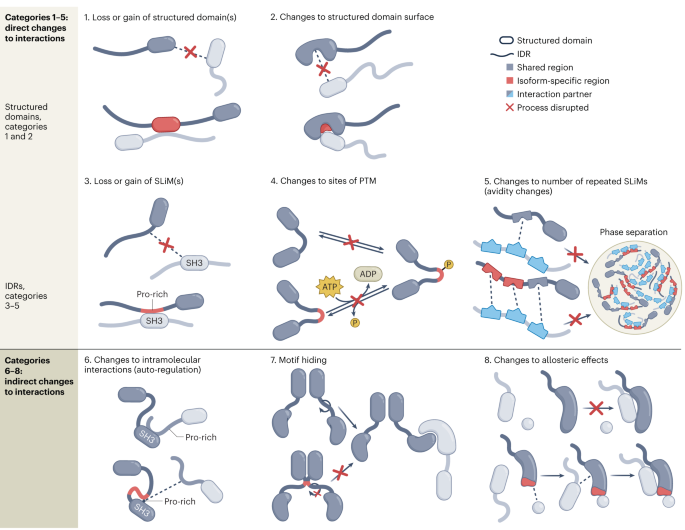
Remember me
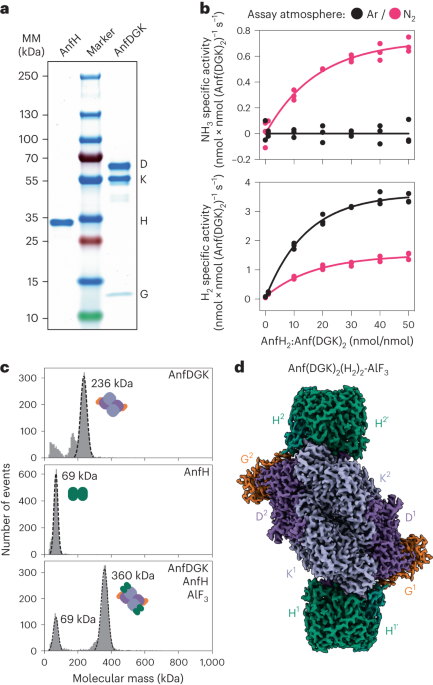

We purified LRRK1(Δ1–19), where the N-terminal 19 residues, predicted to be disordered, were deleted, and imaged it in the presence of Rab7a, ATP and GTP. Most particles classified as monomers, with a small subset forming dimers (Extended Data Fig. 1). The monomer particles yielded a 3.6-Å structure of LRRK1(Δ1–19) (Fig. 1b–d, Table 1 and Extended Data Fig. 1). Although the catalytic C-terminal half of LRRK1, which contains the ROC, COR, kinase and WD40 domains (‘RCKW’), adopts a J-shaped architecture similar to that of LRRK2 (ref. 17), the location of the N-terminal LRR domain differs between LRRK1 and LRRK2 in a functionally important way. The LRR of LRRK2 physically blocks access to the kinase’s active site (Fig. 2a), in what appears to be an autoinhibited conformation16. In contrast, the LRR of LRRK1 is shifted towards its WD40 domain, leaving its kinase’s active site exposed (Figs. 1b and 2a). The only interaction made by the LRR domain with the rest of LRRK1(Δ1–19) is a contact with the kinase’s C-lobe (Fig. 1e). Interestingly, the residue in the kinase involved in this contact corresponds to N2081 in LRRK2, where a mutation linked to Crohn’s disease has been identified11. The kinase domain of LRRK1 is in the open, or inactive, conformation with its DYG motif (a tripeptide involved in ATP binding) ‘out’. Even though the overall conformation of LRRK1(Δ1–19) would not prevent a Rab substrate from being engaged, we did not see any density for Rab7a in our LRRK1 map. This could be explained in part by the presence of an autoinhibitory loop extending from COR-B into the kinase active site, which we discuss below. The ANK domain was not visible in our map.
Table 1 Cryo-EM data collection, refinement and validation statisticsFig. 2: Comparison of monomeric LRRK1 and LRRK2.
a, The LRR domain in LRRK1 does not sterically block access to the kinase’s active site as it does in LRRK2. Models and cartoons are shown for LRRK1 (left), LRRK2 (center) and an overlay of the two structures (right). In the overlay, the ‘RCKW’ portion of LRRK1 and LRRK2 is shown as a surface representation, with the kinase in orange and the remaining domains in gray (light gray for LRRK1, dark gray for LRRK2). The directions of close-ups shown in b–d are indicated on the left-side panel. b–d, These panels show comparisons between LRRK1 (left) and LRRK2 (center) focused on features that are different between the two structures, and a superposition to highlight those differences (right). The insets highlight the region of the structure shown in the main panel. b, The αC helix in LRRK1’s kinase domain is several turns longer than its counterpart in LRRK2. c, LRRK1’s WD40 domain has features that would clash with an element analogous to LRRK2’s latch helix. d, LRRK1’s C-terminal helix is shorter than LRRK2’s.
An unusual feature of LRRK1 is the length of its kinase’s αC helix: it is approximately four turns longer than is typical in kinases (Fig. 2b). The extra residues pack against the COR-B domain through an extensive hydrophobic interaction that is unique to LRRK1 (Extended Data Fig. 2a,b). Interestingly, the RCKW moiety of full-length LRRK1 fits well within a map of LRRK1RCKW (Extended Data Fig. 2c), indicating that the presence of the N-terminal repeats does not alter the conformation of the catalytic half of the protein. This contrasts with LRRK2, where the position of the ROC–COR domains differs significantly between the full-length and LRRK2RCKW structures (Extended Data Fig. 2d). It is possible that the more extensive interface between LRRK1’s αC helix and COR-B domain rigidifies the RCKW portion of LRRK1. Additionally, the longer helix is involved in one of LRRK1’s dimer interfaces, discussed below, which would not be possible with a helix of standard length.
The penultimate propeller blade of LRRK1’s WD40 domain has several distinctive features (Fig. 2c). It contains a ~112 residue disordered loop, unique to LRRK1. The third and fourth strands of this blade are unusually long and would clash with where LRRK2’s hinge helix interacts with the WD40 domain (Fig. 2c); LRRK1 lacks a hinge helix or an equivalent structural element that can interact with the WD40 domain. LRRK2’s hinge helix is inserted at the start of the LRR domain and precedes a ~100 residue disordered loop that contains key phosphorylation sites for binding members of the 14-3-3 family of proteins20, which are involved in regulating signaling in eukaryotic cells. The analogous, much shorter sequence in LRRK1 (residues 244–255) is disordered in our structure.
The C-terminal helix is a structural feature shared between the two LRRK proteins. In LRRK1, the last six residues of the protein are disordered, resulting in a helix that is shorter than that of LRRK2 (Fig. 2d). It was postulated that for LRRK2 the C-terminal helix, kinase N-lobe and COR-B form a regulatory hub where phosphorylation of residue T2524 in the C-terminal helix could regulate kinase activity14. There are no known phosphorylation sites on LRRK1’s C-terminal helix, and it does not extend far enough to contact the kinase N-lobe or COR-B. Since AlphaFold’s21 (https://alphafold.ebi.ac.uk/) predicted LRRK1 structure has a fully folded C-terminal helix, we wanted to understand whether this is a result of AlphaFold’s LRRK1 being modeled in an active state or a more fundamental feature of unknown function. We deleted the last six residues of LRRK1 [LRRK1(Δ2010–2015)] and measured phosphorylation of Rab7a, a LRRK1 substrate8, in cells. We did not observe a significant difference in Rab7a phosphorylation between full-length LRRK1 and LRRK1(Δ2010–2015) (Extended Data Fig. 3), suggesting that the end of the C-terminal helix does not play a major role in LRRK1 regulation. We note that we could not detect LRRK1 in 293T cells in the absence of overexpression (Extended Data Fig. 4a,b), and that no peptides for either LRRK1 or LRRK2 were found in the 293T cell proteome in a previous study22. Similarly, we could not detect statistically significant differences in the expression levels of any of the LRRK1 constructs tested throughout this work (Extended Data Fig. 4c–g).
Cryo-EM structure of dimeric LRRK1We imaged full-length LRRK1 in the presence of guanosine diphosphate (GDP). Surprisingly, this construct yielded almost exclusively dimers (Extended Data Fig. 5), of which we obtained a 4.6-Å structure (Fig. 3, Table 1, Extended Data Fig. 5 and Supplementary Video 1). Each molecule in the dimer has the same conformation we observed in the monomer; however, the ANK domain is fully resolved in the dimer structure.
Fig. 3: Cryo-EM structure of dimeric LRRK1.
a,b, Cryo-EM map (a) and model (b) of dimeric LRRK1. Domains are indicated for the LRRK1a monomer in b.
Surprisingly, despite the similarities in their domain organization and the structures of the monomers, the LRRK1 dimer bears no resemblance to that of LRRK2 (Fig. 4a,b). While LRRK2 forms a parallel dimer mediated by a single homotypic interaction involving its COR-B domain (Fig. 4b), LRRK1 forms an antiparallel dimer mediated by several homo- and heterotypic interactions (Fig. 4a). The ANK–LRR domains of each LRRK1 wrap around those of the other, making symmetrical contacts between the ANK:ANK and LRR:ANK domains (Fig. 4c,d). While the resolution is too low to determine specific interactions at the ANK:LRR interface, the surfaces involved are electrostatically complementary (Fig. 4d). A major interaction interface is formed by the kinase C-lobe and LRR domains of one monomer and the opposite molecule’s ANK domain (Fig. 4e). In contrast to LRRK2, whose dimer is mediated entirely by the COR-B domain in its RCKW moiety, the LRRK1 dimer shows minimal direct interactions between RCKWs; the only contact seen in our structure is a homotypic interaction involving residues 1,263–1,265 in the kinase N-lobe (Fig. 4f).
Fig. 4: Comparison of dimeric LRRK1 and LRRK2.
a,b, Models of LRRK1 (a) and LRRK2 (b) shown in surface representation. The models are shown with the bottom-right monomer in the same orientation to highlight the differences in the architecture of the dimer. The different interfaces involved in forming the LRRK1 (a) and LRRK2 (b) dimers are indicated. In the case of LRRK1, panel letters next to the interface labels refer to the detailed views shown in this figure. Cartoons of the dimers are shown below the models. c, Close-up of symmetric interface formed by the ANK domains. d, The interface formed by the ANK domain of one monomer and the LRR domain of the other monomer brings together surfaces of complementary charge. e, The kinase C-lobe of one monomer interacts both with the LRR domain of the same monomer and the ANK domain of the other monomer. Along with the interaction in g, this anchors the ANK domain on top of the kinase, where it blocks access to the active site. f, Symmetric interaction between the N-lobes of the kinases. This is the only dimeric interface involving only a domain in the C-terminal half of LRRK1 (RCKW). g, The N-lobe of the kinase of one monomer interacts with the ANK domain of the other monomer. Along with the interaction shown in e, this anchors the ANK domain on top of the kinase, where it blocks access to the active site. In c–g, the insets highlight the area of the structure shown in the main panel. Except for d, all other panels show the LRRK1 model inside the cryo-EM map. h, The cryo-EM map of the LRRK1 dimer, colored by domains, is shown in the same orientation as the model in a. i, Rotated view of the dimer map showing how the kinase active site is buried. j,k, An additional rotation of the dimer (j) and clipping of the density in front (k) highlights how the kinase from one monomer is buried under the ANK domain from the other monomer.
The recent structures of dimeric LRRK1 (this work) and LRRK2 (ref. 16) all differ significantly from lower-resolution structures reported earlier19. It remains to be seen whether those earlier structures represent different dimeric forms of LRRK1 and LRRK2.
LRRK1 is sterically autoinhibited in trans in the dimerThe main contact between the two LRRK1 monomers, and the most striking feature of the dimer, involves the ANK domains; each ANK domain contacts the opposite molecule’s kinase on both its N- and C-lobes (Fig. 4h–k), effectively blocking access to the kinase. Our structure suggests that the LRRK1 dimer uses the ANK domain to accomplish, in trans, the steric autoinhibition achieved by the LRR domain in the LRRK2 monomer.
LRRK1’s N-terminus stabilizes the dimerThe LRRK1 (full-length) and LRRK1(Δ1–19) constructs resulted almost exclusively in dimers and monomers, respectively, despite differing only in the presence or absence of 19 residues at the N-terminus. This was particularly surprising as AlphaFold predicts the first ~48 residues of LRRK1 to be disordered (Fig. 5a), and because the first residue we were able to model in the LRRK1 dimer was R51 (Fig. 5b). However, we noted two areas of density in our cryo-EM map, near the junction of the ANK–LRR domains and on top of the ANK:ANK interaction, that were not accounted for by our or the AlphaFold models (Fig. 5c and Supplementary Video 1). We hypothesized that these densities could be accounted for by the extreme N-terminus of LRRK1 stabilizing the autoinhibited dimer. This hypothesis predicted that deleting the N-terminus should destabilize the dimer and thus increase LRRK1 kinase activity. We tested this by measuring phosphorylation of Rab7a in cells. We expressed one of three constructs in 293T cells: full-length LRRK1, a 25-residue N-terminal deletion (LRRK1(Δ1–25)), and a 48-residue deletion (LRRK1(Δ1–48)). In agreement with our hypothesis, both deletions resulted in a ~50% increase in phosphorylation of Rab7a compared to full-length LRRK1 (Fig. 5d and Extended Data Fig. 3). This is comparable to what we observed with the hyperactive kinase mutant LRRK1K746G (equivalent to the PD-linked R1441G mutation in LRRK2) (Fig. 5d and Extended Data Fig. 3). The difference in activity between LRRK1(Δ1–25) and LRRK1(Δ1–48) was not statistically significant, suggesting that the first 25 residues are involved in stabilizing the LRRK1 dimer. To obtain a measure of the Kd for dimerization, we analyzed wild-type (WT), full-length LRRK1 using mass photometry. Based on our analysis, we estimate the Kd for dimerization to be 0.6 μM (Extended Data Fig. 6). This value is almost nine times lower than the concentration we used to prepare cryo-EM grids with full-length LRRK1, in agreement with our observation that most of the protein was found in dimers. Although LRRK1 expression levels may be low, depending on cell type23, LRRK1 has been shown to accumulate near the plasma membrane24, where the locally higher LRRK1 concentrations may drive dimerization.
Fig. 5: Disordered loops in LRRK1’s ANK and WD40 domains help stabilize the autoinhibited dimer.
a, The ANK and LRR domains of the AlphaFold model of human LRRK1 (Q38SD2) shown docked into our cryo-EM map of LRRK1’s dimer. The N-terminal residues 1–48, which are unstructured in the AlphaFold model and not included in our model, are shown in purple. The insets in a–c highlight the area of the structure shown in the main panel. b, Same view as in a with our model of the LRRK1 dimer shown inside the cryo-EM map to highlight that R51 is the first residue modeled in our structure. c, The ANK–ANK interface in the LRRK1 dimer. The purple density corresponds to the region of the cryo-EM map unaccounted for by our current model. R51, where our model begins, is indicated. d, Rab7a phosphorylation in 293T cells expressing full-length LRRK1 or N-terminally truncated constructs missing the first 25 or 48 residues. LRRK1(K746G), which increases Rab7 phosphorylation in cells, and LRRK1(D1409A), which inactivates the kinase, were also tested. 293T cells were transiently transfected with the indicated plasmids encoding FLAG–LRRK1 (WT or mutant) and GFP–Rab7. Thirty-six hours post-transfection the cells were lysed, immunoblotted for phospho-Rab7 (pS72), total GFP–Rab7 and total LRRK1. The mean ± s.e.m. is shown, ****P < 0.0001; NS, not significant; one-way ANOVA with Tukey’s multiple comparisons test. Individual data points represent separate populations of cells obtained across at least three independent experiments (n values shown in EDF 3). e, Weak density (gray arrow) connecting the WD40 domains in our dimer maps. Cartoons above the map indicate the orientation of the maps in e and f and the location of the weak density. f, Cryo-EM map of the LRRK1 dimer with the WD40 domain from the AlphaFold model of LRRK1 docked in; residues 1,792–1,902 were predicted to form a disordered loop. g, Rab7a phosphorylation in 293T cells expressing GFP–Rab7a and full-length WT LRRK1 or a LRRK1 variant missing residues 1,798–1,885 from its WD40 domain. LRRK1(K746G) and LRRK1(D1409A) were used as controls. 293T cells were transiently transfected with the indicated plasmids encoding FLAG–LRRK1 (WT or mutant) and GFP–Rab7. Thirty-six hours post-transfection the cells were lysed, immunoblotted for phospho-Rab7 (pS72), total GFP–Rab7 and total LRRK1. The mean ± s.e.m. is shown. *P = 0.0317, one-way ANOVA with Tukey’s multiple comparisons test. Individual data points represent separate populations of cells obtained across at least three independent experiments (n values shown in EDF 3).
Given these results, we used AlphaFold multimer to model a dimer of the first 25 residues in LRRK1 (Extended Data Fig. 7a). Although the per-residue confidence values are low for the entire 25 residues (as would be expected from AlphaFold’s prediction that the first 48 residues of LRRK1 are disordered), the model shows an interface formed by a short antiparallel β-sheet involving residues 11–15. Interestingly, these residues (and the next one) are the most highly conserved motif at the N-terminus of vertebrate LRRK1s (Extended Data Fig. 7b).
LRRK1’s dimer is stabilized by a loop in the WD40 domainWe noticed a large density in the LRRK1 dimer adjacent to the C-terminal helices and connecting the two WD40 domains (Fig. 5e and Supplementary Video 1); this density was weak and seen only when the map was displayed at lower threshold. We wondered if this was an artifact due to dynamic masking during processing in cryoSPARC25 (https://cryosparc.com/), or to the two-fold symmetry applied to the map of the dimer. We reprocessed the data either using a mask that excluded the region where the density had appeared, or without applying symmetry. In both cases, the unaccounted-for density persisted. We thus wondered if the long LRRK1-specific loop in the WD40 domain (residues 1,791–1,907; Fig. 5f), which we had not been able to model, could be involved in forming the density, and in stabilizing the autoinhibited LRRK1 dimer. We engineered a deletion of most of this loop, LRRK1(Δ1,798–1,885), predicted (by AlphaFold modeling) to maintain proper folding of the WD40 domain, and measured phosphorylation of Rab7a in 293T cells expressing either full-length LRRK1, or the LRRK1(Δ1,798–1,885) construct. In agreement with our hypothesis, deletion of the WD40 loop resulted in a significant increase in Rab7a phosphorylation in cells (Fig. 5g and Extended Data Fig. 3).
A loop from COR-B inhibits LRRK1’s kinaseOur initial model for the kinase domain of LRRK1 showed density in the back pocket of the kinase that was not accounted for by the model. The density was located where Y1410 from the DYG motif would dock in the DYG ‘in’, or active, conformation. Symmetry expansion and focused refinement of the dimer dataset showed that a loop from the COR-B domain (residues 1,048–1,082), which is predicted by AlphaFold to be entirely disordered (Extended Data Fig. 8a,b), threads into the kinase domain active site (Fig. 6a and Supplementary Video 1). The equivalent loop in LRRK2 is half as long and does not extend towards the active site (Extended Data Fig. 8c). The side chain of F1065, at the tip of the loop, sits inside the back pocket of the kinase (Fig. 6b), occupying the position of Y1410 in the DYG-in conformation. A similar ‘plugging’ of the kinase back pocket was observed in the DDR1 kinase (Extended Data Fig. 8d–f)26. This suggests that the COR-B loop is an autoinhibitory element in LRRK1. We tested this by measuring Rab7a phosphorylation in 293T cells expressing either WT LRRK1 or LRRK1(F1065A), which we expected would at least partially relieve the autoinhibition. In agreement with this, the F1065A mutation led to a two-fold increase in the level of Rab7a phosphorylation in cells, comparable to that observed with the hyperactive K746G mutant (Fig. 6c and Extended Data Fig. 3).
Fig. 6: A loop from the COR-B domain directly inhibits LRRK1’s kinase.
a, Close-up of the kinase domain in the cryo-EM map of monomeric LRRK1; the yellow density corresponds to a loop from the COR-B domain that reaches the kinase active site. The inset highlights the area of the structure shown in the main panel. The dashed outline indicates the region shown in b. b, Our model of LRRK1 shown inside the cryo-EM map around the kinase’s active site. The DYG motif, in its ‘out’ conformation, is shown. F1065, a residue in the COR-B inhibitory loop, occupies the kinase’s ‘back pocket’, where Y1410 must dock to bring the DYG motif into its ‘in’, or active, conformation. c, Rab7a phosphorylation in cells expressing full-length LRRK1 WT or carrying a F1065A mutation. LRRK1(K746G) and LRRK1(D1409A) are the same controls used in Fig. 5. 293T cells were transiently transfected with the indicated plasmids encoding for FLAG–LRRK1 (WT or mutant) and GFP–Rab7. Thirty-six hours post-transfection the cells were lysed, immunoblotted for phospho-Rab7 (pS72), total GFP–Rab7 and total LRRK1. The mean ± s.e.m. is shown. ****P < 0.0001, one-way ANOVA. Individual data points represent separate populations of cells obtained across at least three independent experiments (n = 8, except for F1065A, where n = 3). d, Expanded view of the area shown in b, without the cryo-EM map. The three sites of PKC phosphorylation in the COR-B inhibitory loop—S1064, S1074 and T1075—are shown in addition to F1065. e,f, Rab7a phosphorylation in 293T cells expressing GFP–Rab7a and full-length WT LRRK1 or LRRK1 carrying a combination of the F1065A mutation with three phosphomimetic mutations (S1064E/S1074E/T1075E) in the COR-B inhibitory loop (e) or truncated (Δ1–48 and Δ1–25) versions of LRRK1 with or without the phosphomimetic mutations (S1064E/S1074E/T1075E) in the COR-B inhibitory loop (f). The triple phosphomimetic mutant is abbreviated as ‘S/T → E’ in the graphs. LRRK1(K746G) and LRRK1(D1409A) were used as controls. 293T cells were transiently transfected with the indicated plasmids encoding for FLAG–LRRK1 (WT or mutant) and GFP–Rab7. Thirty-six hours post-transfection the cells were lysed, immunoblotted for phospho-Rab7 (pS72), total GFP–Rab7 and total LRRK1. The mean ± s.e.m. is shown. ****P < 0.0001 (P = 0.6730 for Δ1–25 versus Δ1–48). One-way ANOVA. Individual data points represent separate populations of cells obtained across at least three independent experiments (n = 3 (e) and 6 (f)).
LRRK1 contains several consensus sites for phosphorylation by Protein Kinase C (PKC)24. Three of these—S1064, T1074 and S1075—are found in the autoinhibitory COR-B loop (Fig. 6d), and their phosphorylation significantly increases LRRK1’s kinase activity24. In addition, preventing phosphorylation (by mutating the residues to alanine) reduces Rab7a phosphorylation in cells, while phosphomimetic mutations (to glutamate) increase it, although to a lesser extent than phosphorylation24. Our structure provides a mechanistic explanation for this activation: phosphorylation of these residues disrupts the loop, which is nestled against the kinase domain, releasing F1065 from the kinase’s back pocket and allowing the DYG motif to adopt the active ‘in’ conformation. Based on this model, one might expect that combining the F1065A mutation and phosphomimetic mutations in the COR-B loop (S1064E/T1074E/S1075) would not result in further activation, as the F1065A mutation already removes the autoinhibitory interaction. In agreement with this, LRRK1 carrying all four mutations resulted in the same increase in Rab7a phosphorylation in cells as seen with either F1065A or the phosphomimetic mutations (Fig. 6c,e,f and Extended Data Fig. 3).
Our work revealed two separate autoinhibitory mechanisms in LRRK1: (1) autoinhibition by the COR-B loop, present in both the monomer and dimer structures, and (2) steric autoinhibition of the kinase by the ANK domain, which occurs in trans and is dependent on dimerization. Given the seemingly independent nature of these mechanisms, we wondered if their effects would be additive. To test this, we introduced the phosphomimetic mutations in the context of the N-terminal deletions: LRRK1(Δ1–25)(S1064E/S1074E/T1075E) and LRRK1(Δ1–48)(S1064E/S1074E/T1075E). As shown previously24, Rab7a phosphorylation in 293T cells expressing full-length LRRK1 carrying the triple phosphomimetic mutations increased by a factor of 2 relative to WT LRRK1 (Fig. 6f and Extended Data Fig. 3). Combining these mutations with either the 1–25 or 1–48 N-terminal truncation of LRRK1 did not result in a statistically significant increase in Rab7a phosphorylation in cells (Fig. 6f and Extended Data Fig. 3). It remains to be seen whether the phosphomimetic mutants in the COR-B loop disrupt dimerization on their own, thus negating the effect of the N-terminal deletions.
An evolutionary analysis of LRRK1 and LRRK2Structural information on LRRK2 has built up over the last few years14,15,16,17. The data we presented here on LRRK1 allow us to establish the structural signatures that define these two proteins. We set out to analyze the conservation of these features throughout evolution to understand which ones are most likely to be tied to LRRK1- or LRRK2-specific biological functions. We expect that this information will shed light on the etiology of PD and bone diseases.
We began by evaluating the evolutionary origin and phylogenetic distribution of LRRK proteins, defined as those with 40% or higher sequence coverage relative to human LRRK1 or LRRK2, to ensure complete coverage of the ROC, COR-A, COR-B and kinase domains (Methods). We found that LRRK proteins are present in a wide range of metazoan species, and that related proteins are present in amoeba. Phylogenetic analyses of these proteins revealed five distinct and well-supported LRRK clades, with amoeba proteins forming a single clade and the remaining four clades containing only metazoan proteins (Fig. 7a and Extended Data Fig. 9), as has been observed previously27. Notably, arthropod and nematode LRRK proteins, which are annotated as either LRRK1 or LRRK2, are in fact found in a clade (labeled LRRK3 in Fig. 7a) that is distinct from vertebrate LRRK1 and LRRK2.
Fig. 7: Evolution of structural motifs in LRRK proteins.
a, Maximum likelihood phylogenetic tree of LRRK protein homologs (for complete listing of proteins, protein alignment and complete phylogenetic tree, respectively, see Supplementary Datasets 1–3). Shaded clades are LRRK proteins found in metazoans, using nomenclature proposed in ref. 27. The tree is rooted on the only non-metazoan LRRK homologs, found in Amoebozoa, but their validity as the ancestor to metazoan LRRK proteins is not well established28. Asterisks indicate bootstrap branch support (*>75% support, **100% support). b, Expanded views of the phylogenetic tree shown in a, highlighting major metazoan clades that contain members of LRRK1, LRRK2 and LRRK3. Asterisks indicate bootstrap branch support as in a. c, Cartoons of LRRK1 and LRRK2 showing structural features whose conservation was analyzed here. The panels where results are presented for each feature are indicated. d, Schematic of the boundaries used for determining whether a feature shown in c is present in a given LRRK. Amino acid (a.a.) sequences and numbers are shown for regions flanking the sequences whose lengths (e,f,h) or where the presence of key residues in the COR-B autoinhibitory loop (f), or basic patches (g) were measured in our analysis. e, Representative proteins from each metazoan LRRK protein family were sampled from vertebrates (human (Homo sapiens) and frog (Xenopus laevis)), echinoderms (starfish: Asterias rubens), cnidarians (coral: Dendronephthya giganteas), arthropods (fruit fly: Drosophila sechellia) and nematodes (Caenorhabditis elegans). The length of the WD40 loop, as measured between the well-aligning motifs shown in d, is shown next to each homolog. f, As in e, except measuring the length of the COR-B loop between the well-aligning motifs shown in d. Filled boxes indicate the presence of key residues involved in autoinhibition and activation—the Phe that docks into the kinase back pocket (‘F’), and the three phosphorylation sites (‘P1–P3’). g, As in e, except querying for the presence of basic patches. Filled boxes indicate the presence of a basic patch as defined by three basic amino acids in a stretch of four residues between the regions defined in d. h, As in e, except measuring the length of the region containing the αC helix between the well-aligning motifs shown in d.
We next asked when characteristic features of LRRK1 and LRRK2 arose during metazoan LRRK protein family evolution, focusing our evolutionary analysis on four structural features that distinguish LRRK1 from LRRK2: (1) the LRRK1-specific WD40 loop involved in autoinhibition, (2) the LRRK1-specific COR-B loop involved in autoinhibition, (3) the three LRRK2-specific basic patches found in the ROC domain that mediate microtubule binding17, and (4) the length of the αC helix in the kinase’s N-lobe, which is much longer in LRRK1 than in LRRK2 (Fig. 7c,d). We were not able to analyze two other features—the presence of the COR-B:COR-B and WD40:WD40 dimerization interfaces in LRRK2 that are required for the formation of the microtubule-associated filaments, and the differences in length in the C-terminal helix that emerges from the WD40 domain—due to the fact that the protein alignment in these regions was not of sufficient quality to confidently infer relatedness. Consistent with the role that the WD40 loop plays in LRRK1 regulation, we found that this is a conserved feature of metazoan LRRK1s not found in any other metazoan LRRKs (Fig. 7d,e and Extended Data Fig. 10). We also found that an extended (>27 residues) COR-B loop is conserved in LRRK1 proteins (Fig. 7f and Extended Data Fig. 10). Our structure of LRRK1 suggests that the COR-B loop, as defined in Fig. 7d, must be at least 26 residues long for the autoinhibitory mechanism we identified to be possible. Our analysis showed that all three basic patches that mediate microtubule binding are found only in LRRK2s from jawed vertebrates (Fig. 7g and Supplementary Fig. 1). All other LRRK2s we examined had at least the first two basic patches. Finally, the length of the αC helix in the kinase’s N-lobe is a feature found in both LRRK1s and LRRK3s (Fig. 7h).
Comments (0)