
Remember me

Super-enhancers are estimated to contain tenfold more distinct protein factors than typical enhancers and are algorithmically defined using the Rank Ordering of Super-Enhancers (ROSE) script [1,2,3]. Using chromatin immunoprecipitation of active regulatory marks, most commonly Med1, BRD4, or H3K27ac coupled with next-generation sequencing (ChIP-seq), ROSE identifies areas of the epigenome with exceptionally high signal density known as super-enhancers (SEs).
SEs were first discovered in transgenic mice in and termed locus control regions in 1987. Further characterized in murine embryonic stem cells (mESCs), super-enhancers adopted their moniker in 2013 and were observed near transcription factors required for pluripotency, suggesting they may enrich at genes critical for cell identity in the setting of healthy and diseased states [4, 5]. Several genes involved in tumorigenesis and tumor progression were similarly found to be activated by SEs in cancer, notably MYC [2]. Transcriptional regulation by SEs is often critical for downstream gene expression, as CRISPR deletion or interference of the distal SE significantly reduces expression of its target gene. In the case MYC, deletion of its SE in mice results in complete loss of MYC expression in hematopoietic lineages [6].
The etiologies of enhancer changes described in cancer are almost invariably attributed to cell-intrinsic genomic alterations, including (1) activation of oncogenic signaling and, (2) de novo formation of transcription factor (TF) binding sites, as well as (3) focal amplification of non-coding active regulatory regions [7,8,9,10]. Here, we highlight literature on the origins of enhancer and SE reprogramming in cancer, including recent advances in cell-extrinsic SE reprogramming by the tumor microenvironment as well as in extrachromosomal DNA, phase separation, and higher order chromatin structure.
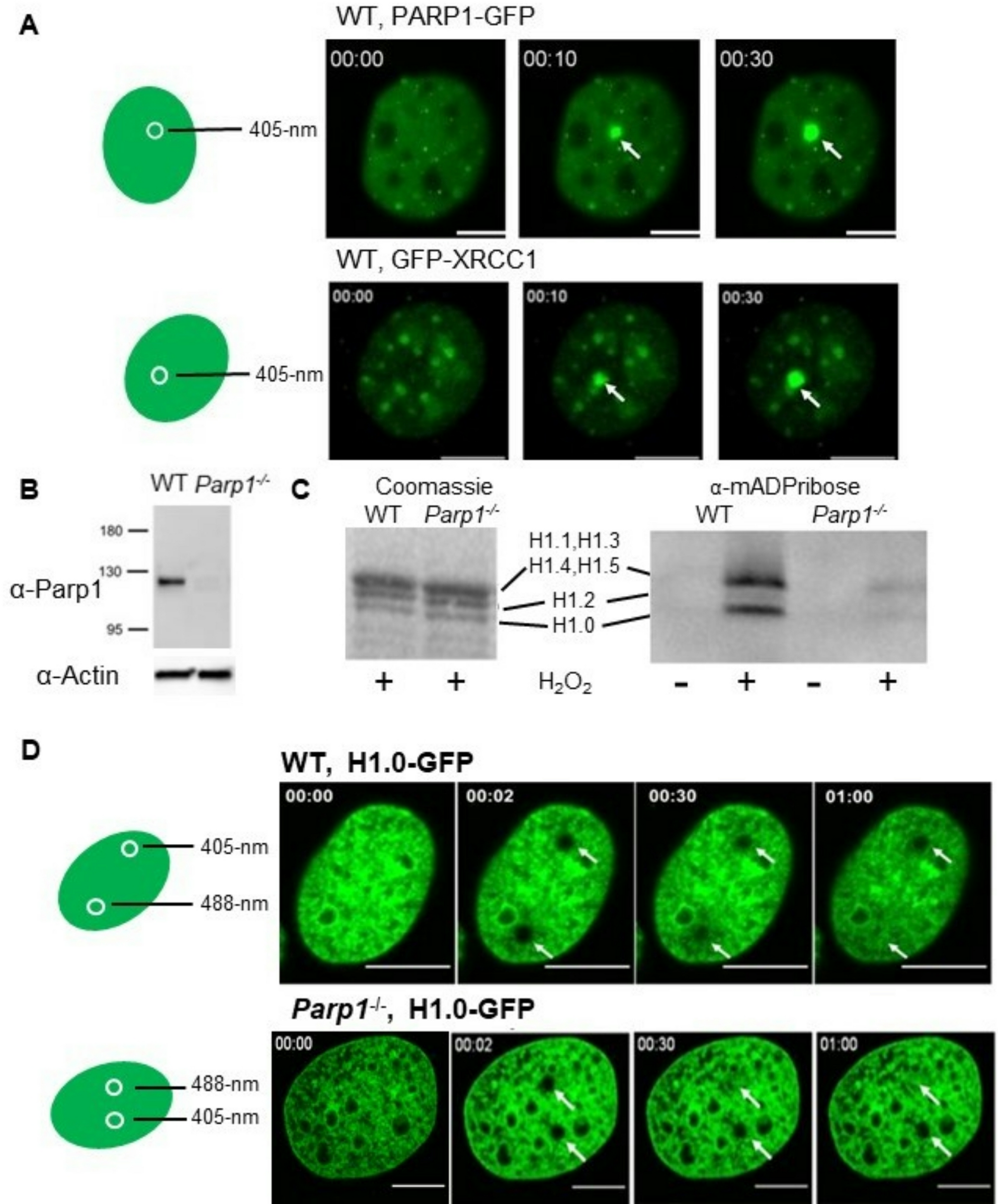
Oncogenic signalingPerhaps the most well-recognized etiology of SE reprogramming in cancer is downstream of somatic coding region mutations that activate oncogenes or inactivate tumor suppressor genes. In renal cell carcinoma, loss of the frequently mutated tumor suppressor VHL that encodes the E3 ligase for the HIF transcription factor, directly results in the formation of numerous aberrant SEs due to HIF accumulation [11]. In sporadic colorectal cancers, ~ 80% of cases are observed to have mutations in the tumor suppressor APC that encodes a member of the β-catenin destruction complex [12]. Accumulated β-catenin subsequently translocates to the nucleus to bind the TCF/LEF family of transcription factors and activate transcription of Wnt target genes including the previously mentioned and known target MYC (Fig. 1) [13]. Indeed, Hnisz et al. observed dense binding of TCF4 within the MYC SE and its motif was enriched among gained SEs in CRC [7]. Thus, oncogenic signaling from somatic mutations shape the super-enhancer landscape in CRC [7].
Fig. 1
Oncogenic signaling results in transcription factor occupancy at gained SEs in cancer. Metagene plots showing TCF4 occupancy (red, left bottom), a transcription factor activated by Wnt signaling, at SEs gained in CRC over normal colon (left top). Metagenes plots showing ERα occupancy (red, right bottom) at SEs gained in ER+ breast cancer over normal breast (right top). Figure reproduced with permission from Elsevier. Please refer to the original publication (Hnisz et al. [7]) for more details and citation
Gain-of-function point mutations in transcription factors can also cause aberrant SE formation. The KLF5 transcription factor, an oncogene implicated in several cancers including CRC, exhibits hotspot E419Q mutations in its DNA binding domain results in over 5000 gained binding sites compared to WT KLF5 including de novo SEs at pro-tumorigenic genes [14]. In line with this finding, lung cancer cells overexpressing KLF5 E419Q exhibited greater proliferation than WT. Transcription factor mutations that create SEs are not exclusive to the DNA binding domain. In lymphoma, the frequently mutated transcriptional activator MEF2B exhibits N-terminal hotspot mutations at regulatory residues [15]. These allow the MEF2B D38V mutant to evades repressor binding and, in turn, bind lymphoma-promoting genes [16].
Inactivating mutations in proteins that directly modify histones can also reprogram SEs, especially proteins governing histone methylation, which are mutated in a variety of cancers [17]. Loss of histone methyltransferase KMT2D (also known as MLL4) in lung cancer results in a global reduction in SE acetylation levels due to inhibition of H3K4 methylation at promoters, including at the transcription factor PER2 which negatively regulates glycolytic genes [18]. Yet, disinhibition of these PER2-dependent glycolytic genes nevertheless sufficiently reprograms the SE landscape of these genes to sustain a metabolic dependency in this subset of lung cancer with increased sensitivity to glycolysis inhibiton [18]. Furthermore, loss of MLL4 also impairs formation of de novo oncogenic SEs by the aberrant expression of HOXA9 transcription factor in acute leukemia [19].
In several cancers, whole genome and whole exome sequencing approaches uncovered high frequency mutations in the SWI/SNF family of chromatin modifying enzymes. The SWI/SNF complex features ATP-ase activity capable of de-stabilizing histone–DNA interactions and thereby regulating chromatin accessibility for transcription factor binding. Thus, mutations in the SWI/SNF family, collectively observed in ~ 20% of all human cancers, can impact the enhancer landscape to varying degrees [20]. The discrepancy in effects is likely dependent on which complex member is lost and evicted and the subsequent specific activity of the remaining complex, as well as the tissue-specific chromatin state it operates within.
For instance, PBRM1, which encodes the BAF180 subunit of SWI/SNF, is frequently lost in renal cell carcinoma (RCC). However, PBRM1 silenced RCC cell lines exhibit little change in open chromatin and H3K27ac landscapes including at SEs [21]. Similarly, almost all pediatric rhabdoid tumors exhibit loss of SWI/SNF core subunit SMARCB1, which encodes the subunit SNF5. SMARCB1 loss in this context decreases SWI/SNF occupancy at typical enhancers (TEs) while maintaining occupancy at SEs, an observation that was reproduced in other cancer cell lines as well [22,23,24].
Conversely, a separate study showed loss of ARID1A, mutated in endometrial carcinoma, preferentially affects SEs over TEs. Wilson et al. observed ARID1A occupancy at SEs over TEs, which exhibited increased H3K27ac signal and open chromatin accessibility following ARID1A deletion resulting in activation of invasion genes [25]. These sites were most significantly co-enriched with the histone acetyl-transferase (HAT) P300, which has known roles in enhancer and SE regulation [25]. Epistasis experiments show that both the hyperacetylation of select SEs and the greater invasive phenotype in endometrial carcinoma observed upon ARID1A deletion is attenuated with either genetic or pharmacologic inhibition of P300, suggesting it is required in the setting of ARID1A loss [25]. The exact mechanism of ARID1A and P300 interplay is not fully understood, but appears to be independent of P300 recruitment as ARID1A deletion did not change significantly change P300 genome-wide occupancy [25]. A follow-up study proposes an alternative mechanism. ChIP-seq studies revealed significant co-localization of ARID1A and the repressive histone variant H3.3, which became depleted following ARID1A loss [26].
Fusion proteins and phase separationThe proximity of constituent enhancers, the density of protein factors, and the level of transcriptional cooperativity led to the hypothesis that SEs exist as membrane-less, phase separated condensates, which have recently emerged as an important driver of protein–protein interactions, especially given the intrinsically disordered domains present among transcription factors, Mediator, and BRD4 (Fig. 2) [3, 32,33,34,35,36,37,38,39]. Indeed, in vitro assays of GFP-tagged intrinsically disordered domains alone, derived from BRD4 or Med1, show condensate formation [40]. The significance of this discovery was in uncovering a structural basis for enabling SE control of gene expression. Liquid condensates exhibit unique aggregation and dispersion properties that appear to specifically associated with SEs over typical enhancers. Microscopy showed DNA–FISH probes against SEs, Med1, and BRD4 to exist as overlapping puncta that could be dispersed with 1,6-hexanediol, which disrupts liquid condensates (Fig. 2) [35, 37, 38, 40]. Notably, puncta dispersion correlated with loss of Med1, BRD4, and RNA polymerase II at SEs. Furthermore, MED1 partitioning recruits RNA polymerase II and its positive regulators while excluding negative regulators [41]. Recent biochemical assays show that this phenomenon, termed selective partitioning, depends on the charge pattern of residues within intrinsically disordered regions [41]. Surprisingly, in vitro droplet assays demonstrate cytotoxic chemotherapy agents, such as cisplatin, also congregate within condensates, with a preference for Med1 [42, 43]. This new area of study provides new mechanistic leads for the selective targeting of SE gene expression in cancer, a sought-after goal since the discovery of SEs.
Fig. 2
Super-enhancers exist as Phase separated liquid condensates. DNA-Fluorescent in situ Hybridization (DNA–FISH) of the Nanog SE shows punctate staining in the mouse embryonic stem cells. Co-immunofluorescence (IF) of super-enhancer transcriptional machinery proteins BRD4 and MED1 also show a punctate staining pattern, suggestive of phase separated liquid condensates. Merged view shows overlap of Nanog DNA–FISH and BRD4 and MED1 IF puncta, suggesting co-occupancy of the same liquid condensate. Figure reproduced with permission from Science. Please refer to the original publication (Sabari et al. [40]) for more details and citation
Oncogenic fusion proteins that create aberrant transcription factors can create de novo SEs to maintain transcriptional dependencies. One recurrently detected fusion in leukemia, NUP98–HOXA9, is enriched for intrinsically disordered domains to form a de novo liquid–liquid phase separated puncta with subsequent oncogenic SE formation detected by ChIP-seq [44, 45]. Notably, fusion proteins with fewer phenylalanine and glycine repeats attenuated phase separation, as did mutating phenylalanine residues to serine [44]. Importantly, these changes also decreased leukemia transformation [44]. Other oncogenic fusion proteins capable of forming and sustaining aberrant SEs include PAX3–FOXO1 in rhabdomyosarcoma, ZFTA–RELA in ependymoma, ETO2–GLIS2 in acute megakaryoblastic leukemia, and TCF3–HLF in acute lymphoblastic leukemia [27,28,29,30]. SEs themselves may even be stitched together in an oncogenic fusion event to form a large hybrid SE, such as C19MC–TTYH1 in embryonal tumors with multi-layered rosettes to promote C19MC onco-miRNA expression [31].
Non-coding mutations and polymorphismsMutations in non-coding regions of the genome are observed in cancer. While most are believed to be passenger events, a few are functionally relevant to cancer cells. Mansour et al. describe a SE in T-ALL that overlapped a recurrent somatic insertion in an intergenic region [8]. This insertion creates a de novo binding site for the transcription factor MYB that resulted in the formation of a SE at this locus (Fig. 3) [7]. The downstream gene, TAL1, was rendered exquisitely sensitive to MYB silencing, suggesting it assumes key regulatory control [8]. As proof-of-principle, CRISPR-cas9 deletion of the somatic insertion collapses the super-enhancer, reduces TAL1 expression, and affects cell viability—one of the first studies to concretely demonstrate somatic mutations in non-coding regions could form enhancers that are inherently oncogenic [8].
Fig. 3
Recurrent non-coding insertions lead to super-enhancer formation and TAL1 expression. Left, H3K27ac ChIP-seq tracks showing a super-enhancer at the locus of TAL1 in Jurkat and MOLT-3 cells, both of T-cell acute lymphoblastic leukemia (T-ALL) origin. This super-enhancer is absent in DND-41 T-cell leukemia cells and fetal thymus cells. Right, non-coding region sequences underlying the TAL1 super-enhancer. Note the recurrent insertions (in red) in Jurkat and MOLT-3 cells, as well as in eight patients with T-ALL, that correlate with formation of the TAL1 super-enhancer. Figure reproduced with permission from Science. Please refer to the original publication (Mansour et al. [8]) for more details and citation
A more systematic process of non-coding somatic mutations occurs in B cell lymphomas. B cells express activation-induced cytidine deaminase (AID) for somatic hypermutation and class switch recombination [46, 47]. However, these can occur at non-immunologic loci that generate translocations and mutations that contribute to B cell lymphoma tumorigenesis. A recent study showed 92% of diffuse large B cell lymphoma (DLBCL) samples, the most common form of lymphoma in the US, exhibit hypermutation and a characteristic AID mutation signature [48]. In 2014, two studies identified AID activity at B cell SEs, with subsequent hypermutation of these non-coding SE regions, including hotspot mutations suggestive of selection at the SE regulating BCL6, a transcription factor that regulates B cell proliferation [46,47,48,49].
In contrast to the Mansour et al. study in T-ALL in which a mutation created a binding site for a transcription factor to form a SE, non-coding mutations in DLBCL occur in existing SEs and alter the binding sequences of transcriptional repressors. This disinhibition results in even greater expression of SE associated oncogenes including BCL6, BCL2, and CXCR4 [48]. At the BCL6 SE, a recurrent mutation prevents binding by the transcriptional repressor BLIMP1 and confers increased fitness in DLBCL cells. When these mutations were corrected back to the WT allele, dropout of DLBCL cells was observed compared to isogenic cells retaining the BCL6 SE mutation [48]. These data show how the process of somatic hypermutation in B cells amplify SE-mediated oncogene expression.
Inborn polymorphisms that affect transcription factor binding also occur in SEs. Oldridge et al. report a G > T polymorphism at the SE of the transcriptional co-regulator and oncogene LMO1 that associated with neuroblastoma susceptibility in GWAS [50]. The G is the reference and risk allele, critical for the GATAA binding motif for the GATA transcription factor within the SE, while the protective alternative allele T breaks this sequence, resulting in decreased GATA occupancy at the LMO1 SE and gene expression [50]. In a European cohort, heterozygous (G/T) and homozygous (T/T) protective allele carriers exhibited significantly increased survival compared to G/G patients [50]. A similar study showed a C > T somatic mutation at an enhancer region of LMO1, suggested to originate from an APOBEC-like cytidine deaminase mutational signature, that created a MYB transcription factor binding site which increased expression and dependency on LMO1 in T-ALL [51]. However, this enhancer was not of sufficient size or density to meet SE criteria [51]. These examples provide further examples of how single nucleotide variations can create or delete transcription factor binding motifs with consequences on SE formation, downstream gene expression, and cancer dependency.
Focal amplificationAmplification is one of the most established etiologies of SE reprogramming in cancer alongside oncogenic signaling. Approximately 25% of neuroblastomas exhibit amplification of the oncogene MYCN which is the strongest correlate for high-risk disease and poor prognosis [52]. While non-amplified MYCN binds to a canonical CACGTG sequence, amplified MYCN results in widespread promiscuous occupation of an ubiquitous CANNTG motif (where N can represent any base) [53]. The result is amplified MYCN binding nearly every promoter and enhancer in the genome in a phenomenon termed “enhancer invasion” that results in globally increased transcription [53]. Dysregulated MYCN is sufficient to form aberrant oncogenic SEs in neuroblastoma [54].
Non-coding regions containing super-enhancers may similarly be amplified over the course of tumorigenesis [55,
Comments (0)