Machine learning encompasses a broad range of data-driven methodologies, including both classical algorithms (e.g., regularized regression, support vector machines, tree-based methods) and more complex approaches such as deep learning. These techniques are particularly well suited for pattern recognition, nonlinear modeling, and predictive analytics in high-dimensional datasets.
In drug development, ML methods have been increasingly applied to molecular screening based on structural and physicochemical similarities to existing compounds, prediction of drug-target interactions, optimization of lead compounds, intelligent drug design, pharmacokinetic and pharmacodynamic modeling, prediction of adverse effects, and identification of novel therapeutic targets (e.g., antibody-based therapies).
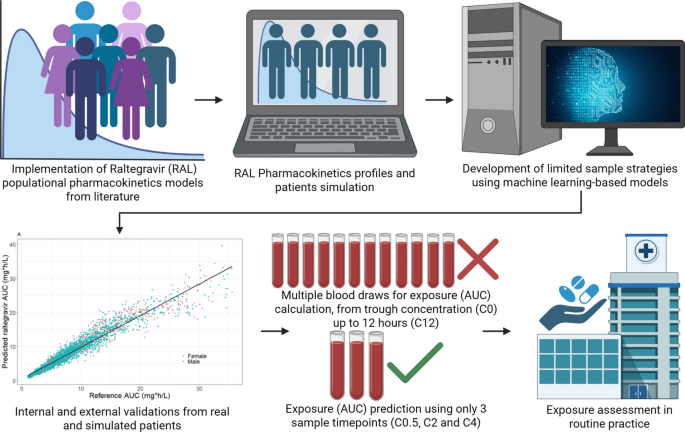
The primary clinical application of this approach lies in therapeutic drug monitoring and exposure assessment in settings where intensive pharmacokinetic sampling is impractical. By reducing the number of required blood samples while maintaining predictive accuracy, ML-based limited sampling strategies may facilitate individualized exposure assessment in special populations, PK/PD studies, and drug-drug interaction evaluations. Although machine learning has broad applications in drug discovery and development, the present work is specifically positioned as a translational tool to enhance clinical pharmacokinetic practice.
Although the general framework of training machine learning models on Monte Carlo simulations derived from population pharmacokinetic models has been previously described for other antimicrobial agents, the present study extends this approach to raltegravir, a drug characterized by high interindividual variability and clinically relevant exposure challenges. By incorporating dual external validation (independent POPPK simulation and real-world postpartum data) and direct comparison with MAP-Bayesian estimation, this work further evaluates the robustness and translational applicability of the ML-based limited sampling strategy framework.
In this study, we developed machine learning models to estimate raltegravir steady-state AUC0–12 using only three sampling time points (C0.5, C2, and C4). Model training relied on extensive Monte Carlo simulations for the generated from the Bukkems et al. (2021) (19) POPPK model. While increasing simulation size may improve apparent performance on simulated data, prior work suggests diminishing returns and potential overfitting to the simulation model when using very large numbers of simulations; in Labriffe et al. (2022) (16), external performance was optimal around ~ 5,000 simulations and worsened for ≥ 10,000. Our model performance was evaluated through both internal procedures and two layers of external validation: one based on an independent POPPK model (25) and another using real clinical data from pregnant and postpartum women (22) (Table I). This design allowed for a rigorous assessment of model robustness in settings that differ structurally and demographically from the training population.
As a result of the feature importance analysis approaches, we obtained the three most important features from each methodology: C0, C2, C3, and C4, with no significant importance observed for fixed predictors such as weight (WT), atazanavir coadministration (ATV), efavirenz coadministration (EFV), low fat diet (LOWFAT), fed status (FED), and pregnancy status (PREG). The ML models were then trained using only combinations of these concentrations (2-by-2, pairs, and 3-by-3, triplets). However, the observed results were unsatisfactory, showing high imprecision and bias.
Including trough concentration (C0) as a predictor improved apparent performance in the simulated datasets, but markedly degraded performance in the external real-world dataset, with ~ 60% bias/imprecision and R2 ≈ 0.40. Because these gains were not reproducible under real clinical conditions, C0-based models were not retained. This divergence suggests that the model may have exploited simulation-specific relationships between C0 and AUC (i.e., patterns driven by the underlying POPPK simulation framework) that did not generalize to real-world data, consistent with overfitting/overparameterization to the simulated training distribution.
Previous work has explored Ctrough (C0) as a marker of efficacy in 800 mg QD regimens, where concentrations above 0.02 mg/L were linked to viral suppression, below 50 copies/mL. However, no reliable cutoff could be established for 400 mg BID dosing (11, 33,34,35). In our analysis, the correlation between Ctrough and AUC0–12 was significant in simulated datasets (R2 = 0.39, Bukkems et al. 2021; R2 = 0.56, Arab-Alameddine et al. 2012), but absent in real patient data (22) (Fig. 7).
Despite the components of the remaining combination (C2, C3 and C4) being initially identified as important features, they likely convey partially overlapping pharmacokinetic information, given their temporal proximity. In particular, C3 lies between C2 and C4 and may not provide sufficiently independent information to meaningfully improve AUC0–12 prediction when combined with adjacent time points. Consequently, combinations restricted to C2, C3, and C4 did not yield robust models and were not retained in the final selection.
Since the results from feature importance analysis were not satisfactory, a comprehensive evaluation of all possible pairs and triplets of concentration measurements of all sampling time collected (C0, C0.5, C1, C2, C4, C6, C8 and C12) was conducted for each ML algorithm (GLMNet, XGBoost, SVM, and Random Forest). We therefore retained C0.5, C2 and C4 as the best-performing and most robust triplet across external validations, while the complete ranking of all combinations is provided in the GITHUB. Our study approach aimed to identify optimal sampling strategies that minimize total patient time for blood collection while reducing overall patient burden focused on sampling within the first 4 h post-dose, avoiding sampling times after that period.
As observed in both simulated pharmacokinetic profiles and real patient data from Moreira et al. (2023) (22), a substantial portion of raltegravir exposure occurs within the early post-dose period. This is consistent with its pharmacokinetic profile, where the apparent terminal half-life is approximately 9 (7,8,9,10,11,12) hours (23, 24), while a shorter distribution (α-phase) half-life of ~ 1 h accounts for much of the total AUC. Consequently, the first hour’s post-dose largely reflect the absorption and early disposition processes that determine overall exposure.
Therefore, sampling within the first 4 h captures the most informative portion of the concentration–time curve, where rapid concentration changes occur, whereas later timepoints mainly reflect slower elimination and contribute less to total exposure. From a machine learning perspective, these early concentrations provide sufficient information for the algorithms to learn the relationship between observed concentrations and AUC0–12, enabling accurate exposure prediction while minimizing patient burden from prolonged blood sampling.
This systematic screening enabled both hyperparameter optimization and robust model training. Across the training dataset, ten-fold cross-validation, and the test set, the XGBoost final model maintained excellent predictive performance. XGBoost consistently outperformed the other algorithms, in accordance with previous findings (13, 14, 17, 21, 36). The high concordance between training and test results, with RMSE and MPE values below 10% and 1%, respectively (Table II), further indicates that the model achieved strong predictive accuracy without signs of overfitting.
The sampling combination yielding the best predictive performance was C0.5, C2, and C4. Once this optimal triplet was identified, we examined whether adding clinical covariates or engineered features could further improve prediction. Neither covariate information nor derived predictors, such as concentration differences (C2-C0.5, C4-C2, C4-C0.5) or ratios (C2/C0.5, C4/C2, C4/C0.5), as proposed by Codde et al. (2024) (13) enhanced model accuracy, and therefore they were excluded from the final model. The optimized XGBoost model ultimately retained only C0.5, C2, and C4 as predictive features for estimating raltegravir AUC0–12.
External validation using simulations from the Arab-Alameddine et al. (2012) (25) POPPK model represented a stringent and independent test of generalizability. Despite structural and covariate differences between the two PK models, XGBoost maintained strong performance, with low bias (< 2%) and relative RMSE values under 15%. When validated in real patients from Moreira et al. (2023) (22), predictive accuracy declined, as expected when transitioning from simulated to real-world data, but overall performance remained acceptable (bias 5%, imprecision 24%). The decrease likely reflects sources of variability not represented in the simulated training sets, including physiological changes during pregnancy, variability in adherence, and real residual variability not captured in the near-zero-error simulations.
Although the RMSE observed in the real patient dataset is slightly higher than that observed in simulated datasets, it remains within the range reported in previous studies evaluating ML-based or regression-based LSS models for pharmacokinetic exposure prediction. Importantly, the relatively low bias suggests that the model does not consistently over- or under-predict exposure, which is a key consideration from a clinical perspective. Therefore, while the decrease in predictive accuracy when transitioning from simulated to real-world data warrants caution regarding generalizability, the observed level of prediction error remains within a range that still provide clinically informative estimates of RAL exposure.
Reducing the residual error in simulation-based training, as implemented here, likely contributed to improved prediction quality across all datasets, in agreement with previous reports showing that machine learning models benefit from simulations with minimal unexplained error (17, 20, 21).
To facilitate clinical implementation, we developed a Shiny web application (https://kathleydata.shinyapps.io/app4) that provides real-time AUC0–12 predictions from input drug concentrations. This approach addresses the main limitation of XGBoost models, the absence of a simple mathematical equation, by offering a proof-of-concept interface accessible for clinical use.
The four ML algorithms were chosen to cover complementary modeling philosophies, from linear to nonlinear and ensemble-based methods. GLMNet (29) is a penalized linear regression (lasso/ridge/elastic net) that stabilizes estimates and can perform implicit feature selection. Support Vector Machines (SVM) (30) use kernel functions to capture nonlinear relationships by projecting data into a higher-dimensional space and fitting a maximum-margin regressor. Random Forest (28) is a bagging ensemble of decision trees that improves robustness by averaging multiple bootstrap-trained trees, reducing variance. XGBoost (27) is a gradient-boosting tree ensemble that sequentially adds shallow trees to correct residual errors, often achieving strong performance on structured tabular data and complex nonlinear pattern.
We compared our results with the multiple linear regression LSS proposed by Cattaneo et al. (2012) (10), which relied on up to five sampling points but was not externally validated. Although their study reported low bias and high explanatory power, it was unclear whether these metrics were derived from training or validation data. In contrast, our model was rigorously evaluated using cross-validation. When we applied Cattaneo’s five-point equation to our external datasets, its performance was comparable to ours in simulated profiles but deteriorated substantially when tested against real patient data. Reducing the equation to three sampling times further compromised accuracy, with consistently high imprecision across all datasets. Notably, our XGBoost model maintains high predictive accuracy using only three predictors. This may be explained by the nonlinear pharmacokinetic characteristics of RAL, including erratic absorption and potential enterohepatic recirculation (6), which can generate complex concentration–time profiles that are difficult to capture with simpler linear models. Gradient boosting algorithms such as XGBoost iteratively combine multiple weak learners (typically shallow decision trees) to model nonlinear relationships and interactions between predictors. These properties make them particularly suitable for pharmacokinetic datasets characterized by nonlinear dynamics and variability. Moreover, tree-based boosting methods have been shown to perform well with relatively sparse input data, such as those derived from LSS.
In the MAP-BE evaluation, we systematically assessed all triplet combinations of sampling times that were explored in the machine learning framework (i.e., all possible combinations among C0 to C4 within the clinically feasible window). Since the objective of the present study was to identify optimal limited sampling strategies (LSS) for raltegravir exposure prediction, no a priori restriction was imposed on a specific sampling triplet (such as C0.5, C2 and C4) for the MAP-BE comparison (Table II).
The MAP-Bayesian estimation approaches provided an additional benchmark to contrast machine learning with PK model-based individualized predictions. The method derived from the Bukkems et al. (2021) (19) POPPK model performed adequately in one clinical dataset but showed marked loss of accuracy in simulated patients, highlighting limited generalizability. The MAP-BE approach based on the Wang et al. (2011) (32) model performed poorly across all datasets. Rather than replacing POPPK, our ML-LSS provides a pragmatic alternative that can be easier to deploy and may remain robust when real-world concentration–time profiles deviate from parametric model assumptions, as suggested by its superior performance versus MAP-BE in our external validations.
The present approach should not be interpreted as a replacement for population pharmacokinetic modelling. Rather, machine learning models trained on Monte Carlo simulations derived from validated POPPK models represent a complementary strategy. While MAP-BE remains the gold standard for model-based individualized prediction, ML algorithms may capture complex nonlinear relationships between limited sampling points and systemic exposure and may offer improved predictive performance in certain settings. Additionally, once trained, ML models can be implemented without requiring real-time structural model fitting, potentially simplifying clinical application.
One of the main limitations of the present study is the absence of real pharmacokinetic profiles obtained under QD dosing regimens of 800 mg or 1200 mg, which precluded the evaluation of model performance at doses other than 400 mg BID, this limitation may affect the generalizability of the proposed model across different dosing regimens. Future studies should therefore aim to externally validate and refine the model using real-world pharmacokinetic data from alternative raltegravir dosing strategies.
A further limitation is that, while AUC0–12 is a practical and widely used exposure summary for LSS development, it does not fully capture potency-coverage indices (e.g., %T > EC90 or trough/EC90) that may better relate to antiviral activity; future work should assess ML-based LSS targeting these PK/PD metrics directly.
Our XGBoost model was developed using specific populations represented in the POPPK simulations and presented robust performance across training, test, and in one of the external validation datasets encompassing substantial diversity: sex, BMI categories (including obesity), HIV status, pregnancy status, age groups (including elderly), ethnicity (white and black), and varied nutritional conditions. However, populations with substantially different demographic, clinical, or pharmacokinetic characteristics may exhibit lower prediction accuracy, in which its generalizability should be interpreted with caution, underscoring the need for further external validation before broader clinical application, as evidenced by the suboptimal performance observed in the population of pregnant Brazilian women.
Beyond its primary TDM/model-informed precision dosing purpose, this simulation-trained ML-LSS framework could also be applied in clinical development to support sparse sampling design and exposure estimation in trials, provided a sufficiently reliable POPPK model is available.







Comments (0)