Resolving structural variants in pangenome graphs with Swave
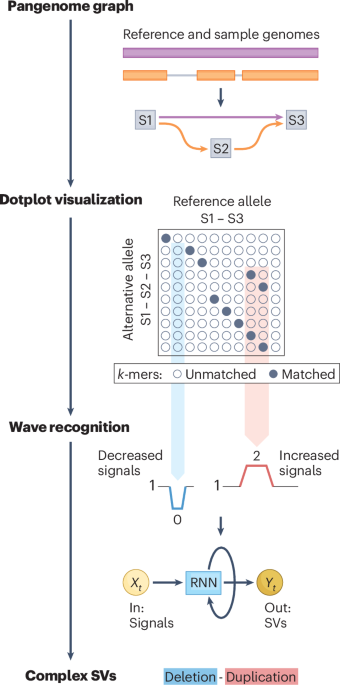
Genome structural variants (SVs) are key drivers of many biological processes. As such, characterizing SVs at the population level provides important insights into species evolution, disease development and so on. The emergence of pangenome technologies offers new opportunities for such analyses. Typically constructed as graph models, pangenomes integrate several genomes into graph ‘snarls’ to encode both shared and specific alleles in the population. These alleles are represented as graph paths traversing nodes, and in principle, their sequence structural differences should be directly observable as SVs. However, in practice, the resolution of current pangenome graphs remains insufficient to fully capture these features. Particularly for complex types or in repetitive regions, pangenome graphs often fail to construct detailed structures of allele sequences within nodes and paths. As a result, existing approaches largely rely on differences in allele length to identify insertions and deletions, which hinders comprehensive discovery of SVs from the population pangenome.
Swave achieved notable advances when applied to both healthy and disease cohorts. In healthy cohorts, it provided a more accurate and comprehensive landscape of population-level SVs through improved genotyping accuracy, precise breakpoint resolution, and the discovery of previously overlooked complex SVs in evolution-associated genes. Notably, Swave reported complex inversion polymorphisms composed of three distinct patterns (balanced, flanked and scarred inversions) that coexisted at the same genomic locus but exhibited distinct frequencies across individuals.


Comments (0)