
Remember me

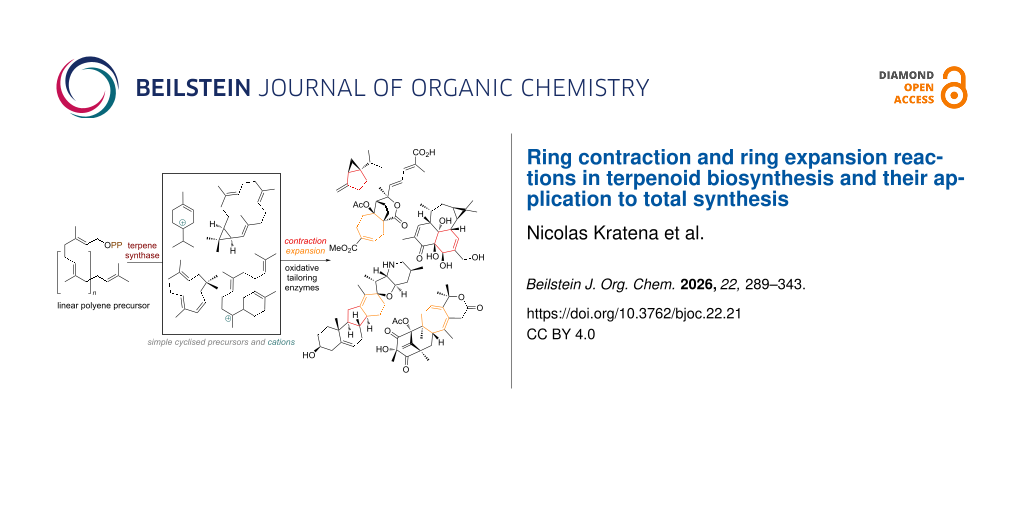

The vast richness of structural diversity in terpenoid natural products has fascinated organic chemists for more than a century now . With more sophisticated techniques for isolation and characterisation emerging over the decades, thousands of closely related compounds could be identified. Among these, the more structurally interesting, rearranged or highly oxidised members can often only be vaguely traced back to their biogenetic origin based on co-isolation and biochemical intuition. Synthetic approaches can sometimes help to solve these puzzling questions or even result in reassignment of the molecular structure . Nevertheless, terpenoids with novel or highly uncommon carbon skeleta continue to attract interest from both biologists and chemists, as they often possess interesting biological properties owing to their unique ring systems, high degree of 3-dimensionality in their structures and oxidation patterns. As obtaining a detailed understanding of a biosynthetic pathway is a dauntingly complex and very labour-intensive process, knowledge about the precise origin of most rearranged terpenoids cannot be secured. Organic synthetic chemistry can help to fill gaps or evaluate, support or revise initially implausible proposed biogenesis routes by attempting to mimic these transformations (= bioinspired or biomimetic synthesis) . In general, the biogenesis of unique carbon skeleton terpenoids can be broken down to several stages. First, a linear polyolefin precursor containing multiples of C5 will be assembled. In general, the cyclisation precursor will interact with a cyclase or synthase enzyme and form a “primary reactive species” which undergoes programmed termination to deliver the most common terpene frameworks. In some instances, these enzymes can also effect ring contraction or expansion directly during the initial cyclisation mechanism . In many cases though, the ring-altering reaction instead takes place at a later stage of biosynthesis, when the oxidation state of the terpenoids is being adjusted . Starting with an already substantial number of these common polycyclic frameworks and adding the almost unlimited variability for oxidative enzymatic C–H functionalisation, it is not surprising that terpenoids with completely unique carbon connectivity and ring systems are still discovered every single year.
In this review article, intriguing examples of apparent ring contraction or expansion reactions of carbocycles in terpenoids, both in biosynthesis and application of similar tactics in the total synthesis, are gathered. Our definition includes both radical and polar ring-size altering reactions, transannular cyclisations of macrocycles and cation-mediated rearrangements where fitting. Goal of this review is to gather and compare mechanistic proposals for the biogenesis of ring-size-altered terpenoids and highlight the utility of strategically including such a step in a natural product synthesis.
To start, an overview of the different classes of enzymes, and thus common mechanisms, of ring-size-altering reactions for terpenes will be presented. In Scheme 1 the four most important manifolds for terpenoid modification in nature are depicted.
![[1860-5397-22-21-i1]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Mechanistic overview of enzymes involved in ring-size-altering reactions: A: Difference in ionisation mechanism in terpene synthases; B: General mechanism and summarised catalytic cycle of CYP450 oxidations; C: Summarised catalytic cycle of α-KG dependent dioxygenases, example of gibberellin acid biosynthesis; D: Importance of methyltransferases and halogenases in terpene biosynthesis, example of methylation of botryococcene (5).
Terpene synthase enzymes are centrally important for determining the carbon skeleton and thus family the terpenoid belongs to. These cyclisation enzymes are divided into Class I (active site cleft contains multiple Mg2+-binding motifs which are responsible for the activation of the diphosphate group and ionisation) and Class II (ionisation by olefin or epoxide protonation via a carboxylic acid in the active site) and their reactions thus are mediated by carbocations and olefins, through careful preorganisation of the linear substrate. Alkyl and hydride shifts, stepwise or dyotropic rearrangements have been described to occur during terpene cyclisation, including multiple ring-size modifications.
The CYP450s play a central role for the oxidative functionalisation of terpenes, enabling the rich diversity of secondary metabolites . They contain an iron-protoporphyrin core with a cysteine ligand in the axial position, which cycles through different oxidation states (Fe3+/Fe2+/Fe4+) to form highly reactive oxo species which are effective at abstracting closely positioned aliphatic hydrogens via HAT (hydrogen atom transfer) . The resulting alkyl radicals are often rebound to yield terpene alcohols or epoxides , but other follow-up reactions are possible.
Next, the class of α-KG (ketoglutarate)-dependent dioxygenases also plays an important role, especially for terpenoids, and is therefore included . These enzymes exhibit distinctly different Fe-containing active sites, typically consisting of two histidines, a carboxylate ligand (e.g., aspartate) and, of course, the ketoglutarate which is bound both via the C-2 ketone and C-1 carboxylate to form 1 (Scheme 1C). Upon its coordination with oxygen via 2 the ketal structure 3 can decarboxylate , generating another high-energy ferryl-oxo species 4 (bound with succinate) which can lead to hydroxylation or carbocation chemistry .
Finally, other classes of tailoring enzymes can also act as electrophiles and lead to ring-size modifications via carbocation formation and 1,2-shifts or are otherwise important in their biosynthesis (cf. below for botryococcene (5), Scheme 1D). Methyltransferases, which contain a closely bound S-adenosylmethionine (SAM) as methylating agent in the active site , and halogenases (e.g., vanadium-catalysed HOX synthesis, see Scheme 1D) are of importance in that regard. Apart from these reactions, spontaneous non-enzymatic reactions can also be responsible for triggering ring-size change in terpenoids.
Review Examples of biosynthetic ring-size adjustments Ring contractions and expansions of terpenoids with secured biosynthetic routesThe large family of C10-terpenes (monoterpenes) includes many linear compounds but also mono- and bicyclic systems. The variety of structures arises primarily through complex, enzymatically guided polyene cyclisation events of linear precursors, such as geranyl pyrophosphate (GPP, 6). As the goal of this review is to cover mainly skeletal modifications arising from follow up biosynthetic reactions after cyclisation, only selected examples of ring-size altering fundamental steps in polyene cyclisation mechanisms will be presented now. One such example is the formation of the bicyclo[3.1.0]hexane system present in thujane monoterpenes (see Scheme 2A). Starting from geranyl pyrophosphate (6), monoterpene cyclases first build up a 6-membered ring with an exocyclic carbocation, commonly referred to as α-terpinyl cation 6a. From there, a 1,2-hydride shift gives rise to the isomeric terpin-4-yl cation 6b. By way of cyclopropane formation, a different, so called, thujyl cation 6c is conceivable. Elimination then furnishes the ring-contracted 5/3-ring systems from the original cyclohexyl intermediate to give sabinene (7) and α-thujene (8).
![[1860-5397-22-21-i2]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: A: Ring contraction through involvement of carbocationic intermediates in thujane monoterpene biosynthesis; B: Spirocyclisation of a decalin system, initiated from germacrene A (10) during biosynthesis of the sesquiterpenoid β-vetivone B (11); C: Decalin-system ring contraction in pleuromutilin (13) biosynthesis through multiple 1,2-hydride and alkyl shifts.
Another example of a 6→5 ring contraction can be found in the family of spirocyclic C15-sesquiterpenes, such as β-vetivone B (11, see Scheme 2B) . The linear precursor farnesyl pyrophosphate (9) is first cyclised by germacrene A synthase (GAS) to its name-bearing product 10. From here, protonation by vetispiradiene synthase (HVS) gives the eudesmane cation 10a, which is a common intermediate in the biosynthesis of bicyclic sesquiterpenes, e.g., aristocholenes. In this particular enzymatic reaction, the 1,2-alkyl shift of a Wagner–Meerwein rearrangement is responsible for building up the spirocyclic carbon framework and ring contraction (10b). From here, follow up oxidations and olefin isomerisation afford β-vetivone B (11), a constituent of aromatic vetiver oil.
Finally, in the biosynthesis of the antibiotic pleuromutilin (13) a similar ring contraction takes place (see Scheme 2C). Cyclisation is initiated via protonation of geranylgeranyl pyrophosphate (GGPP, 12) at the terminal olefin with CpPS (= Clitopilus passeckeranis pleuromutilin synthase), forming the decalin system 12a. A cascade of 1,2-shifts delivers the carbocation 12b which undergoes ring contraction and elimination to give 12c. From here, a second cyclisation towards 12d, 1,5-hydride shift to secondary carbocation 12e and follow-up oxidative decoration of 12f affords the pleuromutilin molecule 13.
A more recently unveiled example of a specific cyclase enzyme effecting a change in ring size during polyene cyclisation was found in pseudolaric acid B (14) biosynthesis . In this case, the linear precursor 12 is first cyclised to give intermediate 14a akin to the α-terpinyl cation (see Scheme 3). From here, quantum chemical calculations indicate that the subsequent 1,2-alkyl shift and olefin cyclisation occur in a single, concerted step. This concerted mechanism allows the reaction to bypass a high-energy, non-stabilised secondary carbocation intermediate that would result from a stepwise migration. This pathway is facilitated by the enzyme's active site, where aromatic residues (e.g., Tyr564) are proposed to provide the essential carbocation stabilisation (likely via cation–π interactions) and thus facilitate alkyl migration. The resulting 5,7-bicyclic carbocation 14b is further elaborated and decorated by selective oxidation reactions to build up the family of pseudolaric acids. For example, after a second ring expansion, and additional 5-ring cyclisation by the pendant alkene a new tricyclic carbocation 14c is formed. From here a 1,2-alkyl shift delivers the 7,6,6-ring system of lydicene (15) . Alternatively, a 1,5-hydride shift affords the secondary carbocation 14d which undergoes ring contraction to afford the cyclopropyl-containing natural product neoverrucosanol (16) .
![[1860-5397-22-21-i3]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Examples of concerted ring expansions of carbocation intermediates in PxaTPS8-catalysed cyclisations towards fungal diterpenoids in nature and transannular cyclopropyl cyclisation.
Another closely examined example of a similar ring expansion was documented by Abe et al. (see Scheme 4) to occur during the biosynthesis of astellifadiene (17), catalysed by the cyclase EvAS (= Emericella variecolor astellifadiene synthase) . Geranylfarnesyl pyrophosphate (18) is first cyclised through 1,11- and 10,14-connections to the tertiary carbocation 18a, and undergoes the previously described ring expansion, cyclopentane cyclisation (analogous to Scheme 3, see above) to 18b. Elimination affords the triene intermediate 19, followed, once again, by transannular cyclisation to give the tertiary carbocation 19a. A subsequent second ring expansion affords the bridged system cation 19b, which is quenched by a 1,5-hydride shift to finally provide astellifadiene (17).
![[1860-5397-22-21-i4]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Sequential ring expansions during astellifadiene (17) synthesis reported by Abe and co-workers.
A distinct and intricate cyclisation cascade was elucidated by Dickschat and co-workers in 2023, describing the biosynthesis of the spirocyclic diterpene spiroluchuene A (20, see Scheme 5), catalysed by the synthase AlTS (Aspergillus luchuensis terpene synthase) . Geranylgeranyl diphosphate (GGPP, 12) is initially cyclised through the well-known 1,10-closure to the cation 20a, followed by a sequence of hydride (20b) and proton shifts to cation 20c, and a second hydride shift to 20d. Subsequent cyclobutane formation via 20e and a ring expansion furnishes cation 20f, which is quenched by deprotonation with cyclopropanation to afford the key neutral intermediate luchudiene (21). One of the olefins in 21 is then reactivated by re-protonation, initiating a second cyclisation sequence that proceeds via cations 21a and 21b. Finally, a remarkable sacrificial carbocyclisation (rupture of the cyclopropane ring) at an aliphatic centre then generates cation 21c, which is quenched by deprotonation to yield spiroluchuene A (20).
![[1860-5397-22-21-i5]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Cyclobutane ring expansion and sequential ring contractions catalysed by the synthase AITS in the biosynthesis of spiroluchuene A (20).
An illustrative example of a complex cyclisation cascade with multiple ring-size modifications was documented by Dickschat et al. during the biosynthesis of the saturated sesterterpene subrutilane (22, see Scheme 6), catalysed by the cyclase SrS (= Streptomyces subrutilus synthase) . Geranylfarnesyl diphosphate (18) is first cyclised through 1,11- and 10,14-connections to the tertiary carbocation 22a, which undergoes ring expansion (22b) and a 14,18-cyclisation to 22c. A 1,5-hydride shift from the macrocycle generates cation 22d. Subsequent 2,9- and 3,7-cyclisations afford the key carbocation intermediate 22f, which is finally quenched by a deprotonation with concurrent cyclopropanation, effectively contracting a cyclopentane to a cyclobutane, to yield subrutilane (22).
![[1860-5397-22-21-i6]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Ring expansion and transannular ring contraction of a cyclopentane to cyclobutane in the biosynthesis of the sesterterpene subrutilane (22) catalysed by SrS.
Based on quantum chemical studies by Tantillo and Hong the biosynthesis of kaurene diterpenes includes interesting, concerted alkyl migration steps . Starting from GGPP (12) the class II terpene synthase CPS (= ent-Copalyl diphosphate synthase) catalyses decalin formation through cationic polyene cyclisation of ent-copalyl diphosphate (23) (Scheme 7). From here, different enzymes (e.g., ent-Kaurene synthase) can effect cyclisation of cation 23a to the pimarenyl cation 23b, from which earlier works proposed a stepwise cyclisation and alkyl shift to occur. The secondary carbocations which are invoked in this process were found to not be the likely operational intermediates, as calculations showed instead a concerted rearrangement towards the tertiary cation 23c to be more likely. From here, ent-kaurene (24) is obtained directly after elimination. The formation of ent-atiserene (25) involves a more dramatic rearrangement to reach the tertiary carbocation 23d. A triple asynchronous shift occurs, consisting of C12-alkyl (13→16), 1,3-hydride (12→13), and C-13 alkyl shift (16→12). This complex, concerted process directly converts the tertiary cation 23c into the tertiary cation 23d completely avoiding secondary cations and resulting in another 5→6 ring expansion to the bridged system of 25.
![[1860-5397-22-21-i7]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Computationally elucidated concerted cyclisations/alkyl/hydride shifts during the biosynthesis of the terpenes ent-atiserene (25) and ent-kaurene (24).
An early example of a sesquiterpene cyclisation sequence with a ring contraction was documented by Cane et al. during the investigation of epi-isozizaene (26, Scheme 8), catalysed by the synthase EIZS (= Epi-isozizaene synthase) . Farnesyl diphosphate (9) is first ionised and isomerised to (3R)-nerolidyl diphosphate (26a), which undergoes cyclisation to form the bisabolyl cation (26b). A subsequent 1,2-hydride shift yields cation 26c which undergoes spirocyclisation to generate the acorenyl cation (26d). This key intermediate then undergoes a sequence of further cyclisation (26e) and crucial ring contraction via 1,2-alkyl shift to 26f. Methyl migration and quenching by elimination finally afford epi-isozizaene (26).
![[1860-5397-22-21-i8]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Cyclisation events and 6→5-ring contraction during the construction of epi-isozizaene (26) catalysed by EIZS.
Investigative efforts into actinomycetes biosynthesis by Dickschat et al. through deuterium labelling revealed another example for a 4→5 ring expansion which is depicted in Scheme 9 . Intermediate 27 is obtained like shown before (see above, Scheme 4 and Scheme 6), through the enzyme cattleyene synthase (CyS). Next, a concerted ring expansion/ring contraction and additional 2,10-cyclisation delivers the macrocyclic cation 27a. A further 3,6-cyclisation forms the cyclobutane intermediate 27b which can undergo ring expansion via dyotropic rearrangement to give the tetracyclic system of 27c, followed by 1,2-methyl migration (27d) and elimination to furnish cattleyene (28).
![[1860-5397-22-21-i9]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Transannular cyclisations and 4→5-membered ring expansion through dyotropic 1,2-rearrangement of alkyl and methyl groups in the biosynthesis of cattleyene (28).
During the investigation of botrydial biosynthesis the groups of Collado, Cane and Viaud could identify a protein titled BcBOT2 (= Botrytiscinerea BOTrydial) with similarity to microbial terpene synthases . Starting from farnesyl pyrophosphate (9) BcBOT2 catalyses a rare, formal [2 + 2] cycloaddition to form the cyclobutyl carbenium ion 29a which undergoes spontaneous ring expansion via 1,2-alkyl shift (see Scheme 10). The secondary carbocation on the 5-membered ring in 29b undergoes additional cyclisation (and ring contraction from a 9-membered ring to a 6/5 bicycle) to give tertiary cation 29c. From here a 1,2-hydride shift leads to intermediate 29d (presilphiperfolan-8-yl cation), a precursor which was invoked to be involved in the biosynthesis of various sesquiterpenes. Two noteworthy examples are given here: either of the two 1,2-alkyl shifts of the cyclohexane accomplishes ring contraction. Following the blue arrow in the structure of 29d (Scheme 10) a second 1,2-methyl migration is required to furnish silphiperfolene (29) . If instead, the bond indicated with the green arrow migrates the triquinane skeleton is assembled. Notably, Yan reported recently that the sesquiterpene α-terrecyclene (30) is formed through the same carbocation 29d, according to the mechanistic proposal by Coates . By interception of the cation with water we arrive at presilphiperfolan-8β-ol (31)
![[1860-5397-22-21-i10]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Ring expansion in presilphiperfolan-8b-ol (31) biosynthesis and ring contraction of the presilphiperfolan-8-yl cation (29d) towards different sesquiterpene skeleta.
Dickschat and co-workers reported an example for a ring contraction during the biosynthesis of sodorifen (32) . The cyclisation is triggered by the C-methyltransferase SodC (= pre-sodorifen synthase) which catalyses methyl cation transfer from SAM towards the terminal olefin in 9, a rare event in terpene cyclisation chemistry, resulting in 6,11-ring closure (32a, see Scheme 11). From here, a dyotropic rearrangement can give cation 32c directly, alternatively the mechanism could be enabled by deprotonation through an active-site base (32b) and re-protonation of the cyclopropane moiety (32c). A 1,2-hydride shift, followed by a 1,2-methyl shift towards that same carbon and an elimination, forms the tetrasubstituted double bond in 33, the precursor for sodorifen (32), which is furnished by the class I synthase SodD.
![[1860-5397-22-21-i11]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Ring contraction via transannular cyclopropanation and opening of cyclopropane in the biosynthesis of the microbial terpene sodorifen (32).
A well-studied example of an oxidative 6→5-membered ring contraction in terpenoid biosynthesis can be found in the formation of the gibberellin family . In this instance, the terpene ent-kaurene (34, see Scheme 12) is being oxidised both at one of the methyl groups residing at C-4 and the C-7 methylene to ent-7α-hydroxykaurenoic acid (34a). The hydroxy group in 34a can further engage with a CYP450 enzyme at C-6 (different CYP isoforms responsible in different genii) to form an alkyl radical 34b which upon further SET forms an intermediate carbocation 34c, which collapses under an 1,2-alkyl shift to reveal the key intermediate in gibberellin synthesis, gibberellin A12 aldehyde (35). Alternatively, the ent-kaurene core is known to be likely oxidised at C-1 by a thus far unknown biosynthetic oxidation to deliver a secondary carbocation 35a, which can undergo tandem ring expansion/contraction from the 6,6 to the 7,5 system (35b). After elimination the precursor 36 to the large family of grayanotoxin natural products is reached (grayanotoxin II (37) is depicted exemplarily).
![[1860-5397-22-21-i12]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: The crucial CYP450-catalysed oxidative rearrangement defining the skeleton in gibberellin biosynthesis and selective C-1 oxidation and ring expansion/contraction towards grayanotoxin II (37).
Another well-studied example of a CYP450-catalysed oxidation triggering a change in ring size was found to occur during the biosynthesis of some brassicicene natural products such as brassicicene I (38) . The 5-8-5 ring system present in these natural products (see Scheme 13) is transformed into a 5/9/5-bridged system through a methylene C-12–H oxidation mediated by the enzyme BscF, triggering a Wagner–Meerwein rearrangement after radical-polar crossover (38a to 38b) and elimination at the bridgehead methyl, resulting in the exo-olefin. The final intermediate 38c is further elaborated by enzymatic oxidation to give brassicicene C (39) from brassicicene I (38).
![[1860-5397-22-21-i13]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: CYP450-mediated oxidation of cyclopentane methylene expanding the 8-membered ring in the biosynthesis of brassicicene natural products.
In the biosynthesis of aridacin A (40), the initial 6,10-membered bicyclic product 41, after AriE-catalysed polyene cyclisation engages in a HAT with the CYP450 enzyme AriF, abstracting a hydrogen from the C-20 methyl group on the macrocycle . The resulting allylic radical 41a can cyclise directly, subsequently giving a different tertiary radical which can get oxidised and quenched by elimination (not depicted in Scheme 14). Alternatively, a SET can occur directly on the methyl radical, delivering allylic carbocation 41b, which cyclises barrierless to give tertiary carbocation 41c. Regioselective, endocyclic elimination delivers the unoxidised terpene precursor 42 towards aridacins.
![[1860-5397-22-21-i14]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: CYP450-mediated oxidation of an exocyclic methyl group to effect transannular cyclisation across the 10-membered macrocycle towards aridacin A (40).
In the rich family of Euphorbia diterpenoids various ring systems are conjectured to be biosynthetically related, such as the casbane, lathyrane, tigliane, and ingenane skeletal . The transannular aldol reaction of an oxidised casbene (43, produced from GGPP by casbene synthase, CS) product 44 has been studied closely by the groups of Graham and Hamberger who investigated the BGC present in Jatropha curcas and Euphorbia lathyris, respectively (see Scheme 15) . They found that two CYP450 enzymes, CYP71 and CYP726 were primarily responsible for C-4, C-5 and C-8 oxidation of casbene 43 to form intermediate 44 which can isomerise by means of keto–enol tautomerism to give 44a. An additional keto–enol tautomerism allows for the transient formation of 44b, a new enol engaging in an aldol addition towards the highly electrophilic α-diketone moiety. With this, the bicyclic lathyrane skeleton of jolkinol C (45) is assembled selectively from casbene.
![[1860-5397-22-21-i15]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Non-enzymatic transannular aldol reaction enables the formation of the 5/13/3-tricyclic ring system present in lathyrane diterpenoids from the casbane skeleton.
The large family of indole meroterpenoids also contains an interesting, enzyme-mediated ring-expansion reaction. According to the detailed research of their biosynthesis carried out by Oikawa et al. the terpenoid penitrem A (46) is formed from PC-M4 (47, see Scheme 16A). This precursor, exhibiting a 5-membered ring annulated onto the indole core, reacts in a HAT reaction under mediation of PtmK, an enzyme possessing an iron(IV)–oxo metal centre, to deliver the methyl-centred radical 47a. From here, the 5→6 ring expansion takes place via radical mechanism giving rise to the stabilised tertiary radical 47b. Oxidation of this radical to the corresponding carbocation and elimination leads to secopenitrem D (48) directly. From here another oxy-cyclisation and aromatic chlorination leads to the most complex member of the indole meroterpenoids, penitrem A (46).
![[1860-5397-22-21-i16]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: A: Oxidative ring expansion of a cyclopentane by incorporation of a methyl group in the biosynthesis of penitrem A (46); B: Ring expansion and rearrangement of the indole moiety in the biosynthesis of sespenine (50).
Another example of a ring expansion reaction, in this case of the aromatic 5-membered ring of an indole, was discovered to be operational in the biosynthetic pathway towards xiamycins and dixiamycins (see Scheme 16B) . The rearrangements commence by oxidation of C-3 in the indole ring of indosespene (49) by the enzyme XiaF, giving rise to iminium intermediate 49a. Attack of this iminium by the pendant exocyclic olefin affords carbocation 49b, which is quenched by a 1,4-alkyl shift of the aromatic system, and establishment of a C–O double bond at C-12 to give sespenine (50) as the product.
In meroterpenoid biosynthesis, the transformation of andilesin C (51) towards the anditomin skeleton, as investigated by Abe and co-workers, is also an interesting example for ring expansion . The PhyH-like dioxygenase AndF abstracts a hydrogen from C-11 of 51 giving rise to secondary alkyl radical 51a (see Scheme 17). An iron(IV)-hydroxy-mediated rebound then results in the hydroxylation of this position. Upon E1 elimination of alcohol 51b, carbocation 51c is generated and undergoes a 1,2-alkyl shift of the ketone moiety, forming a new bridged 7-membered ring (51d), and completing the skeletal adjustment. A final elimination to the exocyclic olefin delivers the complex, caged structure of anditomin (52).
![[1860-5397-22-21-i17]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Rearrangement and ring expansion in the construction of the complex bridged carbon framework of anditomin (52), mediated by AndF.
Apart from this D-ring expansion of 3,5-dimethylorsellinic acid (DMOA)-derived meroterpenoids, Abe and co-workers were also able to show enzymatically controlled pathways for the B-ring expansion and A-ring spirocyclisation and contraction . From the common precursor preaustinoid A1 (53) a HAT step mediated by the iron(IV)-oxo enzyme PrhA generates a radical (53a) at C-1 of the terpenoid framework (see Scheme 18), resulting in cyclopropanation to give radical 53b. Subsequent cyclopropane opening leads to the ring-expanded B-ring centred, tertiary radical 53c. Finally, abstraction of the allylic hydrogen in this product by an iron(III)-species affords the 6/7/6/6 bridged framework of berkeleydione (54). A different iron(IV)-oxo enzyme AusE is also capable of selectively abstracting the 5α-hydrogen atom, giving rise to alkyl radical 53d, which gets hydroxylated via rebound to give intermediate 53e . Carbocation formation at C-5, followed by a 1,2-alkyl shift and elimination thus furnishes the sister natural product preaustinoid A3 (55), thought to be the precursor to the highly oxidised meroterpenoid acetoxydehydroaustin (56).
![[1860-5397-22-21-i18]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Ketoglutarate-mediated oxidations of preaustinoid A1 (53) en route to complex meroterpenoids, B-ring expansion via radical skeleton re-shuffling, A-ring contraction via Wagner–Meerwein spirocyclisation.
Other interesting, proposed ring-size changing reactions in terpenoid biosynthesisMoving on from these carefully characterised biosynthetic transformations, where often single steps were carried out with specific enzymes or isotope-labelled substrates to prove origin, we now move to the much larger body of speculative biosynthetic relationships. From macrocyclic lathyrane frameworks such as jolkinol C (45), the formation of complex polycyclic tiglianes and ingenanes is also invoked, even though detailed studies have still not been performed . The required C-8→C-14 cyclisation would require a C-nucleophile to be present at C-14 (next to the cyclopropyl group), attacking the C-8 ketone (e.g., in 45). While the topological relation of lathyranes and tiglianes is immediately apparent, the mechanism and required pre-functionalisation for the 8-14 cyclisation are not trivial and have thus far not been supported by extensive synthetic model studies . Instead, an alternative biosynthetic pathway towards tiglianes from a partly Z-configured casbene precursor 57 was proposed and is depicted in Scheme 19 . Crucially after the formation of the 5/13/3-tricyclic system in 58 as before, an oxidation next to the cyclopropane would allow 1,2-migration of the cyclopropyl system after nucleophilic attack by one of the olefins in 59. After a 1,2-hydride shift (59a to 59b) and nucleophilic capture of the tertiary cation to give 60, the full skeleton of tiglianes (e.g., phorbol (61)) would be assembled.
![[1860-5397-22-21-i19]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Proposed putative biosynthetic formation of the tigliane skeleton from an E,E,Z-triene.
An early example of a puzzling ring contraction observed in natural product chemistry was the transformation of santonin (62, see Scheme 20) into the ring-contracted 5-membered compounds lumisantonin (63) and the 5,7-membered ring-expanded guaiane system 64 . The mechanism for this photochemical transformation involves transformation of 62 to a diradical at the unsaturated ketone (62a), cyclisation to form an intermediary highly substituted cyclopropane (62b) followed by single-electron transfer to the zwitterionic 62c and rearrangement to either the dense connectivity of lumisantonin (63), or carbocation capture by solvolysis of intermediate 64a to afford isophotosantonins 64. The closely related guaiane natural products are not believed to be synthesised in nature via this mechanism and instead are formed by different termination events during polyene cyclisation of a key intermediate . Due to the early discovery of santonin in 1830 and the correct elucidation of its structure almost 100 years later, this example with negligible biosynthetic relevance is also included.
![[1860-5397-22-21-i20]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Photocatalytic tandem ring expansion/contraction of santonin to give photosantonin products and guaiane carbon skeleta.
The proposed biosynthesis of stelleroids features an α-ketol rearrangement leading to a 7→6 ring contraction (see Scheme 21A). After a protonation-induced cyclisation of the bicyclic precursor 65 to the bridged system 65a, and exhaustive oxidation to give intermediate 65b, the structure of stelleroid B (66) is reached by a final 1,2-alkyl shift. The kaurene-derived product crokonoid A (68) was traced back to its co-isolated compound crokonoid B (67) by oxidation, carbocation formation and 1,2-alkyl shift in a semi-pinacol rearrangement . The authors of the isolation report proposed an epoxide 67a as initiating species, but a diol could also serve in this function (see Scheme 21B). Upon formation of the secondary carbocation 67b, a 1,2-alkyl migration is invoked, giving rise to the bridged polycyclic system of 68.
![[1860-5397-22-21-i21]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: A: Proposed biosynthesis of stelleroid B (66) from stelleranoid I (65) by ketol rearrangement; B: oxidation and ring expansion in the biogenesis of crokonoid A (68) from crokonoid B (67).
Apart from these larger classes of sesquiterpenoids there are many singular examples of rearranged and ring-size-modified kaurene derivates, such as pierisketone B (69) and pierisketolide A (70). These 7,5,6,5-systems were proposed by the group responsible for their isolation to be built up through oxidation of precursor 71 at C-5 and C-6 to give an epoxide 72, which could undergo semi-pinacol rearrangement to the 7-membered ketone 73. From here, a simple follow up oxidation could give both compounds (see Scheme 22A).
![[1860-5397-22-21-i22]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Singular examples of A,B-ring contractions and expansions in the biosynthesis of sesquiterpenoids euphomilones A (77) and B (78), euphnerin B (76), pierisketone B (69) and pierisketolide A (70).
Alternatively, one can imagine an exhaustively oxidised intermediate like 74 undergoing α-ketol rearrangement to directly deliver the hydroxy group at C-6 (acid 75) for intramolecular lactonisation towards pierisketolide A (70). In the biogenesis of euphnerin B (76) and euphomilones A (77) and B (78) the tricyclic precursor 79 was invoked (see Scheme 22B) . This compound could undergo oxidation at the alkene (and C-1) to give intermediate 80, which after oxidative cleavage affords the enolate of diketone 82 which can undergo transannular aldol addition to give either product 77 or 78 after proton exchange. The same intermediate diketone could be theoretically obtained directly from singlet-oxygen-mediated oxidation of the olefin through hydroperoxide 81 and Hock rearrangement . Finally, euphnerin B (76) is furnished after 1,2-alkyl shift in 83 towards the C-5 carbocation, to afford the 5,6-spirocycle.
The transformation of these 5/7/6/3-membered systems into the ingenane skeleton was exploited to great effect in multiple independent total syntheses of these molecules (vide infra). It is conceivable that an analogous process, starting with oxidation at the methyl group to form an allylic carbocation, 1,2-hydride shift and finally semi-pinacol rearrangement is also responsible for the re-shuffling of the carbon skeleton in nature. While the connection of casbanes, lathyranes, tiglianes, ingenanes and even jatrophanes has been described and was the subject of previous studies, no detailed studies on the transformations connecting all these families have been performed thus far . Novel Euphorbia diterpenoids recently isolated by Wang, Zheng and Yu in 2024 (see Scheme 23A) could be considered “missing links” offering an explanation as to how the strained inside-outside bridgehead of ingenol (85) is formed . Precursor compounds such as euphebranane D (84) could, upon protonation at the exocyclic olefin give a stabilised carbocation 84a/84b. This would allow for the 1,2-alkyl shift and ring expansion to take place, giving ingenol (85). From there, the related ring-expanded diterpenoid euphebranane A (86) was proposedly obtained by 1,2-alkyl shift of 85a to give carbocation 85b and finally 86 by elimination to the diene and epimerisation of the hydroxy group. An alternative mechanistic rationale for the expansion/contraction reaction from the 5/7/7/3-ring system towards a 6/6/7/3-system was proposed by Qiu, Zhou and Yue et al. in the isolation reports of pepluanols A–D . Here, a retro-aldol reaction was invoked to cleave the C–C bond rupturing the 5- and 7-membered ring. The C-4 ketone can then enolise in a way that enables selective C-4 to C-14 ketone aldolisation furnishing pepluanol D (87). The same authors also proposed a putative biosynthetic origin for the intriguing 5/4/7/3-ring system of pepluacetal (89) from pepluanol A (88) by olefin isomerisation (88a to 88b) and 1,6-conjugate addition to build up the new 4-membered ring (see Scheme 23B). Finally, acetal formation on the novel carbaldehyde furnished the structure of pepluacetal (89).
![[1860-5397-22-21-i23]](https://www.beilstein-journals.org/bjoc/content/inline/1860-5397-22-21-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: A: plausible proposed biosynthetic pathway for the tigliane/ingenane skeletal rearrangement and 1,2-shift ring contraction/expansion towards 6/6/7/3 diterpenoids euphebranane A (86) and pepluanol D (87); B: proposed biosynthetic pathway from pepluanol A (88) to give pepluacetal (89).
An interesting example for both the ring expansion and contraction of benzene rings in nature was unveiled during investigations into the biosynthesis of xenovulene A (90, Scheme 24A) . The terpenoid precursor 91 and the polyketide precursor 92 are merged to give rise to the tricyclic system of 93. Oxidation of the electron-rich aromatic system in
Comments (0)