Giant cell tumor of bone is an overall benign but locally aggressive neoplasm, which is defined as the proliferation of mononuclear stromal cells with multinucleated osteoclast-like giant cells scattered within the neoplasm. They are considered histologically benign, but GCTB has a marked tendency toward local recurrence, and in rare cases may metastasize, mostly to the lungs [1, 5].
GCTB accounts for approximately 4–5% of all primary bone tumors and 20% of all benign bone tumors [6, 7]. The principal demographic affected by GCTB is young adults between 20 and 40 years of age, with a slight female predominance, occurring primarily in the epiphyseal region of long bones - most commonly around the knee; GCTB can also occur in the distal radius and sacrum [8].
There is a clear link in the pathogenesis of GCTB to the H3-3 A gene changes, which encodes histone H3.3. The vast majority (approximately 92%) of all GCTBs have a mutation in codon 34, usually the p.Gly34Trp (G34W) [3, 4]. This mutation only happens in the neoplastic mononuclear stroma cells that are believed to be the neoplastic component of GCTB. The G34W mutation is believed to affect histone methylation patterns, thus modifying transcription and causing RANKL overexpression to promote osteoclastogenesis. The multinucleated giant cells are believed to be reactive from the monocyte–macrophage lineage cells, which arose from this signaling [9]. The H3-3 A gene that encodes the histone protein H3.3 acts as an oncogenic driver for developmental and transcriptional mechanisms. Canonical hotspot mutations arise at lysine 28 and glycine 34, with the G34 codon, specifically, the G34W substitution having the strongest correlation for giant cell tumor of bone [3].
There are also rare mutations such as the H3F3B alterations that have been reported, however, they are generally not related and may be related to specific anatomic locations [10]. The H3-3 A mutation has clinical significance since it is absent in histologic mimics such as aneurysmal bone cyst and chondroblastoma. Additionally, Gomes et al. 2014, found that H3-3 A p.Gly34 TRP, was not present in aggressive CGCG of the jaw and giant cell lesions of patients with cherubism [11].
Through next generation sequencing we also identified a pathogenic FANCA p.R951 variant. FANCA is a tumor suppressor gene that encodes the protein Fanconi anemia complementation group A, which is a member of the Fanconi anemia core complex that assembles at damaged chromatin and activates the DNA response by coordinating BRCA 1/2 proteins. Mutations of FANCA are the most prevalent, making up 65% of the heritable Fanconi’s anemia cases that are a cancer predisposition syndrome with elevated risk for a variety of malignant neoplasms [12].
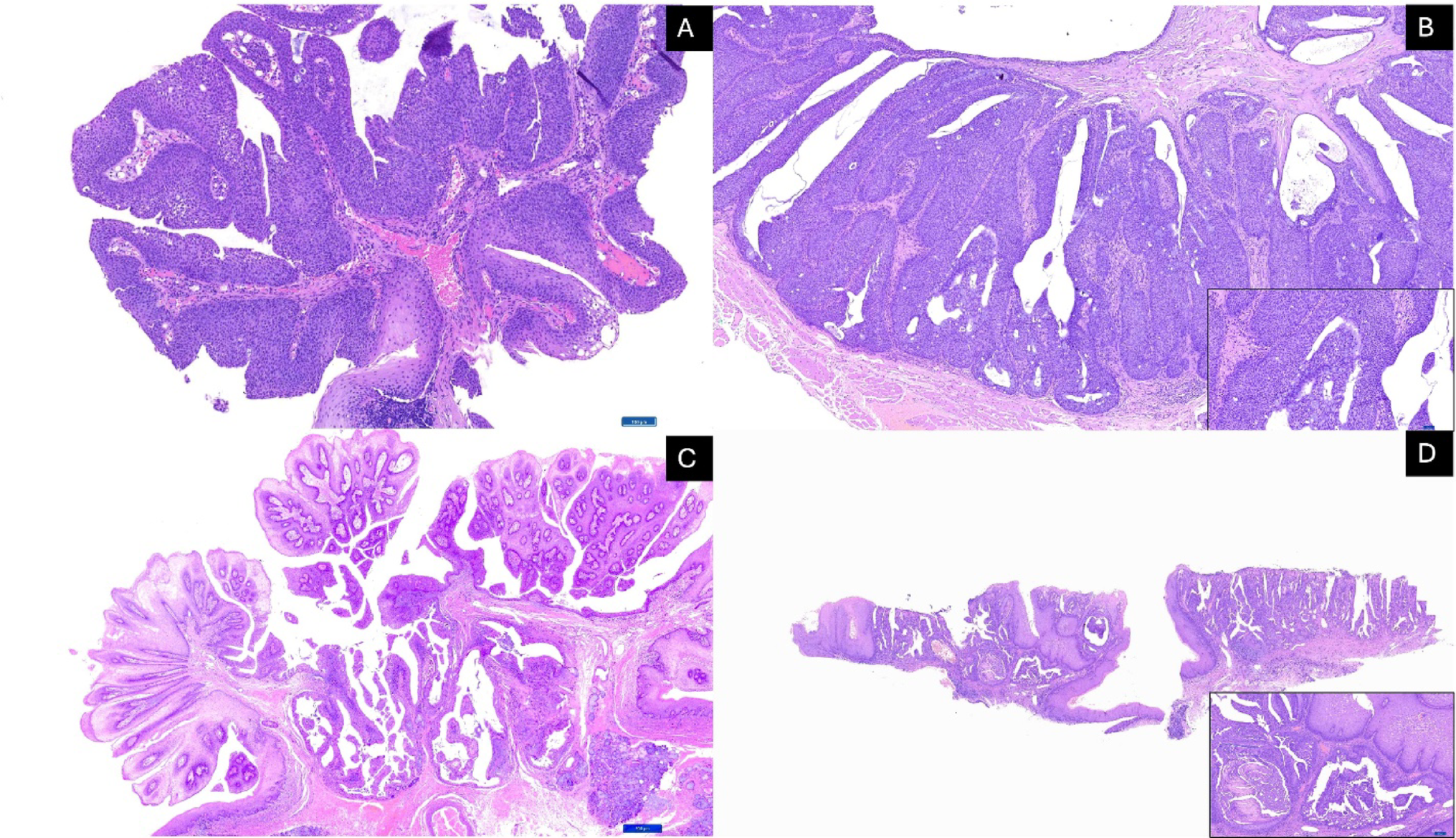
GCTB consists histologically of three main cell types. The main cells are mononuclear stromal cells, the neoplastic cell population; these appear as round to oval cells with vesicular nuclei, moderate eosinophilic cytoplasm, and the occasional nucleolus. Multinucleated osteoclast-like giant cells, which can contain 20 to 50 + nuclei, are spaced randomly throughout the lesion and often clustered around hemorrhage. Finally, there are mononuclear histiocytic cells which are part of the reactive component and tend to be fewer than the stromal cells [13]. Mitotic activity is noted but atypical mitotic figures are infrequent in conventional GCTB. Areas of hemorrhage, hemosiderin deposition, and necrosis are common. Secondary aneurysmal bone cyst–like changes may be present in ~ 14% of cases [14]. Imunohistochemically the mononuclear stromal cells demonstrate strong nuclear positivity for H3.3 G34W mutation-specific antibody, whereas the giant cells are positive for CD68 [13, 15, 16].
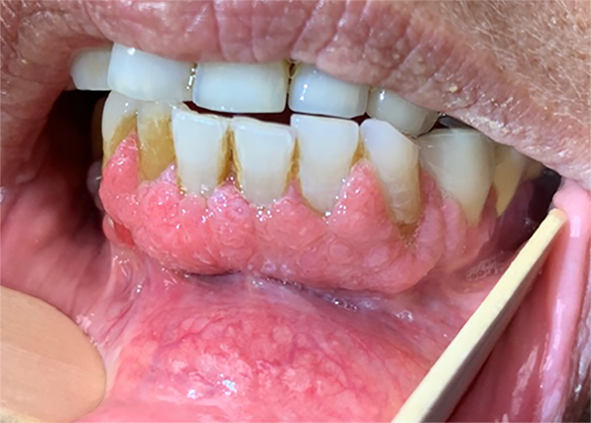
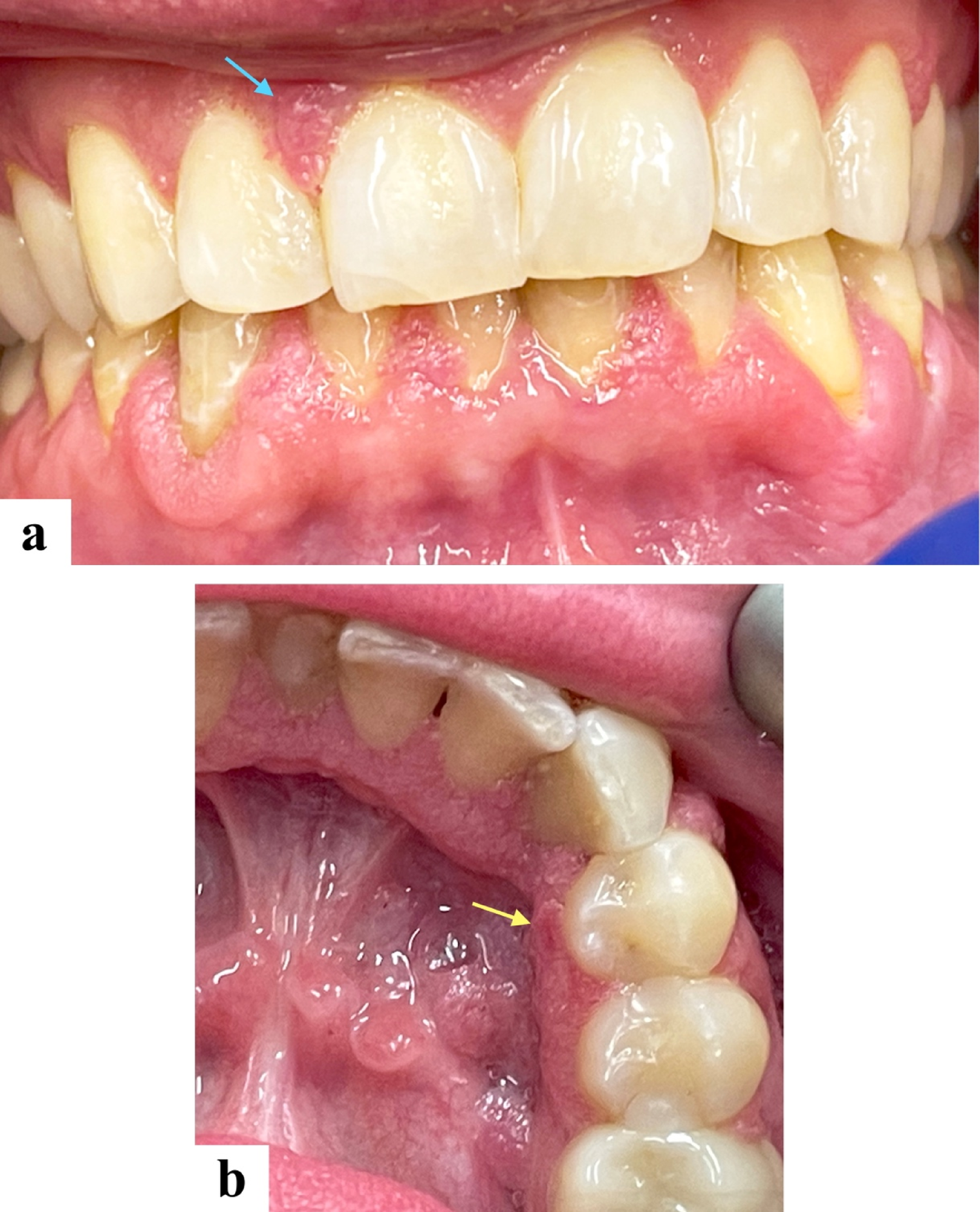
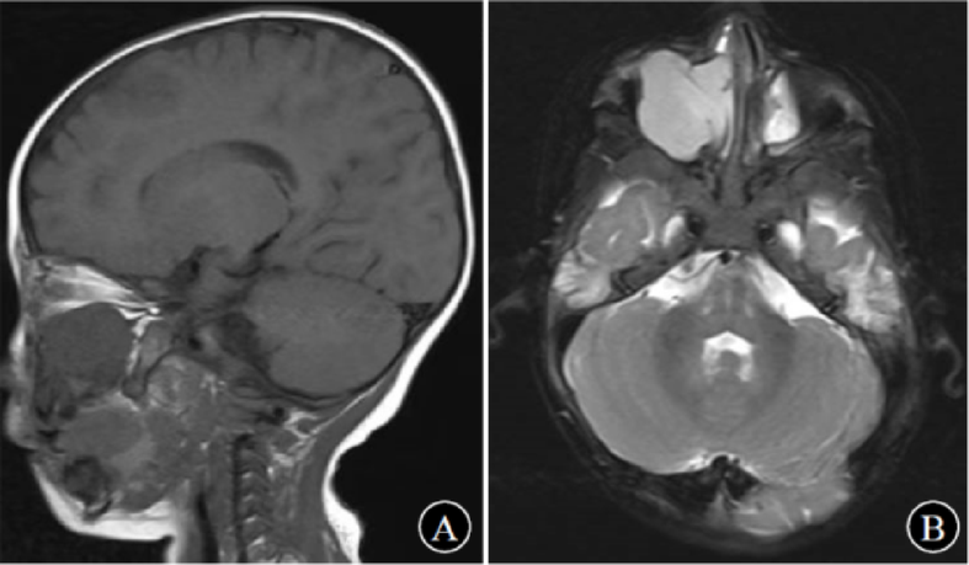
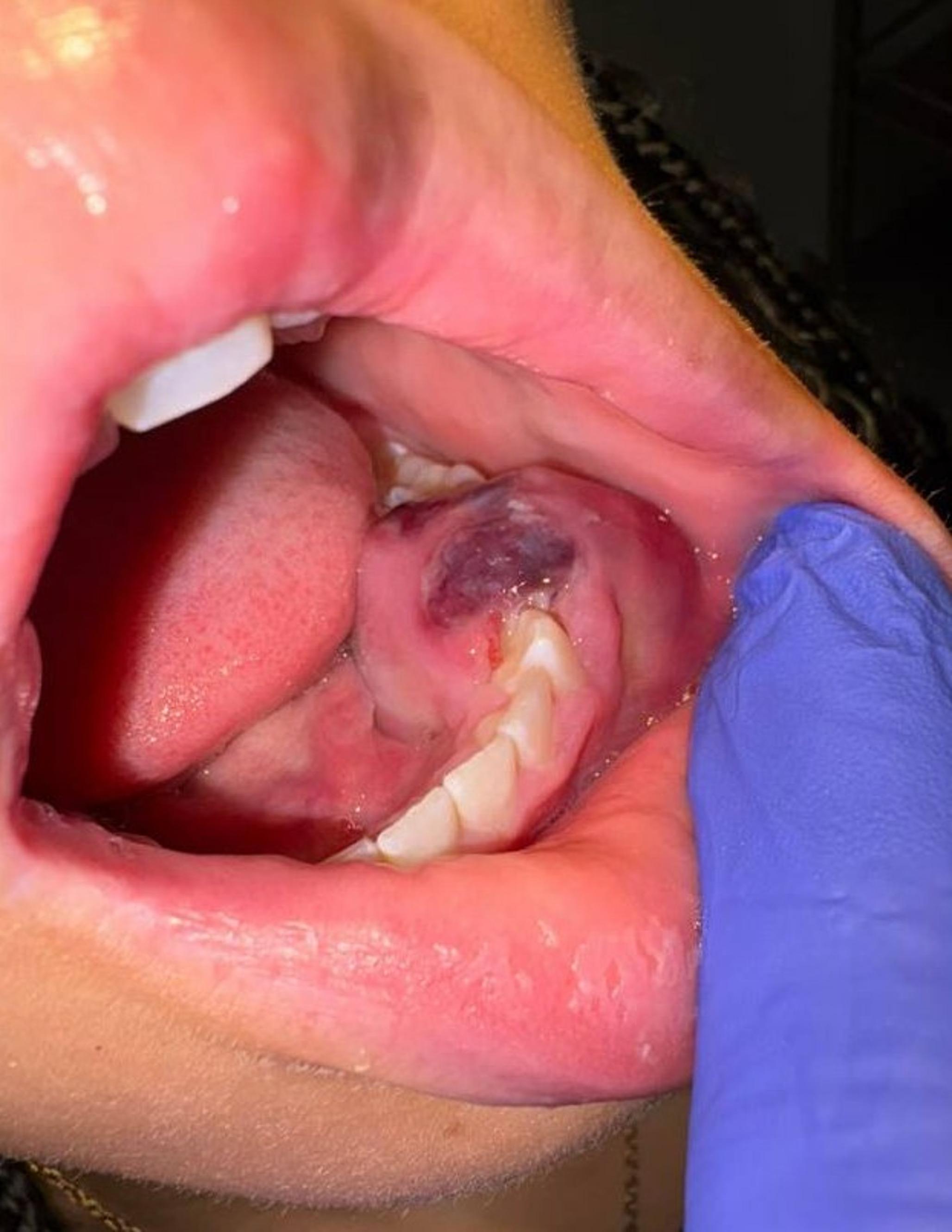
Typical radiographic features include an eccentric lytic lesion affecting the epiphysis and extending to the subchondral bone with non-sclerotic margins, the lack of matrix mineralization, and an appearance similar to the radiographic presentation in our case [17], [18].
Although histologically benign, GCTB has a 20–50% local recurrence rate depending on surgical management [19]. Malignant giant cell tumor of bone is an uncommon aggressive variant that accounts for less than 5% of all GCTB. It can arise de novo (primary malignant GCTB) or more commonly as a secondary transformation from a previously established benign GCTB, generally after multiple recurrences or following prior radiotherapy [20]. Malignant transformation is very rare, typically occurs after radiation therapy, or with recurrent disease. The prognostic factors for recurrence include the site (distal radius and sacrum), inadequate surgical margins, and secondary aneurysmal bone cyst changes [21, 22].
β-human chorionic gonadotropin (β-hCG)–expressing giant cell tumor of bone (GCTB) is incredibly rare. Although the production of β-hCG has been defined for some bone sarcomas, for example, osteosarcoma [23]. However, β-hCG expression in GCTB, in gnathic bones, is exceedingly rare. To date, the only case found in the medical literature described the finding of a GCTB that had changes of an aneurysmal bone cyst-like changes in the head and neck region. In this case, the patient’s serum β-hCG levels returned to normal following resection, confirming that the tumor was the source of hormone production [24]. In our patient, the levels of β-hCG dropped with treatment with denosumab. To the best of our knowledge, this may be the first reported case of β-hCG secreting GCTB in the mandible. Yet, Lawless et al. (2014) published an institutional case series of 40 patients with β-hCG–secreting GCTB [23]. In one case, the tumor arose in the posterior mandible and showed mild positivity for β-hCG on IHC. However, the authors did not clarify whether this patient had elevated serum β-hCG levels and noted that, in several cases, there was a discrepancy between immunohistochemical expression and serum findings. A full summary of reported GCTB in the head and neck region is presented in Table 2.
There is a gap in the literature regarding the prognostic and therapeutic implications of β-hCG expression in GCTB. No systematic studies have been published that investigate the impact of β-hCG positivity on recurrence risk, metastatic risk, or therapy response in these tumors [24,25,26]. Therefore, more research is needed to investigate whether β-hCG expression has clinical relevance other than causing challenges in the diagnostic interpretation. The current standard of care for GCTB remains surgical curettage, either with or without local adjuvant therapy, and denosumab for unresectable or recurrent disease [25, 27]. Though β-hCG expression does not currently influence best treatment practices, recognition of this rare finding is important to ensure the correct diagnosis and highest quality clinical care [23, 24].
Complete surgical excision is the gold standard for GCTB in the mandible, ideally with negative margins to minimize chances of recurrence [27, 28]. Extended intralesional curettage with local adjuvants is preferred, where possible, with recurrence rates up to 27%, depending on technique and adjuvant used. En bloc resection may be required for large or recurrent lesions, but must be weighed against mandibular reconstruction morbidity, particularly in younger patients such as ours [27, 28].
The use of denosumab, a monoclonal antibody to RANKL has changed the management of unresectable or recurrent GCTB28. Denosumab reduces the number of giant cells and induces bone formation. While long-term follow up and recurrence rates after discontinuation remain under assessment [9, 16].



Comments (0)