The results from this study demonstrate that monoclonal expansion of J∆NI8 HSV-1 vectors can result in genetic mutations, some of which can significantly alter the functional properties of the vector, in particular its fusogenic potential. These findings align with the known genomic instability of herpesviruses during replication, underscoring the importance of rigorous quality control in vector production to ensure therapeutic safety.
Some batches of HSV-1 replication-defective vectors may alter neuronal physiology
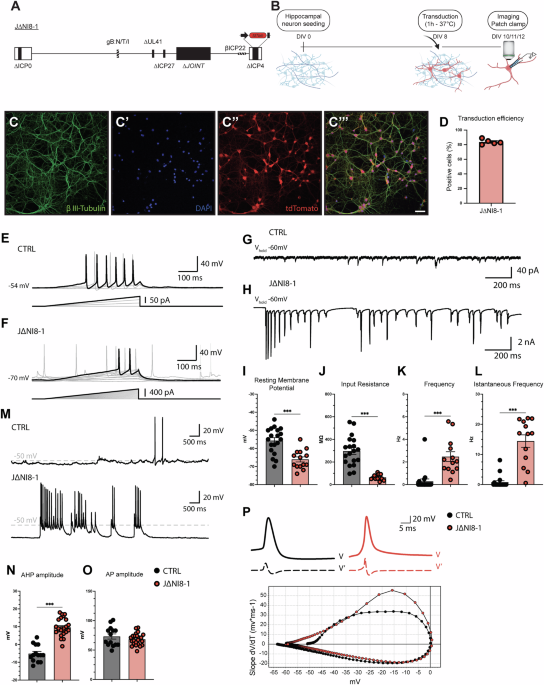
Whole-cell patch-clamp experiments revealed several electrophysiological alterations in neurons transduced with some J∆NI8 vector batches that were not leading to obvious morphological alterations. We examined in detail the effects of one of these batches (J∆NI8-1), and observed increased spiking frequency and bursting activity, reduced Rin, a more hyperpolarized RMP, spontaneous spiking activity (single spike discharges or bursts of action potentials), and altered action potential properties (alterations in the AHP). These alterations are reminiscent of those reported in primary cultures of dissociated sensory neurons infected with syncytial HSV-1 strains, which are thought to be responsible for certain types of neuralgia [12]. Interestingly, despite the authors’ use of a wild-type HSV-1 replication-competent syncytial strain, no marked morphological changes were observed in neuronal cultures in their experimental conditions [12]. Similarly, in our hands neurons appeared viable and displayed no sign of cytotoxicity even at the latest time-point that we employed for patch-clamp experiments (i.e., 96 h after transduction). These data led us to hypothesize that, similar to syncytial HSV-1 strains, some J∆NI8 batches might exert fusogenic effects even if highly deficient and non-replicative. It is likely that the viral particles used to transduce neurons can fuse cells together, rather than induce de novo expression of gB. This hypothesis is supported by previous data on our replication-defective vectors, which show low, if not null, expression of viral genes [22]. Thus, we decided to focus our attention on potentially harmful mutations in the gB gene rather than the de novo expression of the protein.
Genetic mutations and their functional consequences
We then sequenced the DNA of two distinct batches of J∆NI8, one causing the electrophysiological alterations described above (J∆NI8-1) and another (that we named J∆NI8-Syn) that caused clearly detectable fusion in complementing cells during vector preparation and would therefore be routinely discarded. We identified several SNP mutations in these batches, in most of the cases synonymous or intergenic. However, we also identified missense mutations in the genes UL27, UL46, US7, and US10. Of particular interest were the missense mutations in UL27, because this gene codes for gB, one of the most important fusogenic proteins of the Herpesviridae family [33]. The R858H and S869N mutations in gB, observed only in the highly fusogenic J∆NI8-Syn, have established associations with increased membrane fusion activity [34, 35] and are therefore in line with the obvious phenotype of this vector batch. We hypothesized that the other mutations (D285N, A315T and A549I) that are also observed in the J∆NI8-1 batch may not only cause electrophysiological alterations but also favor the fusion of membranes between neurons. In fact, fusion may cause the spreading of excitation and, thereby, hyperexcitability.
We directly addressed this hypothesis in multiple manners. First, we performed a dye coupling experiment and found that Neurobiotin was able to diffuse from the patch pipette and label not only the patched cell but also neighboring tdTomato-positive neurons. Second, we performed dual cell patch-clamp experiments of tdTomato-positive neurons and found that J∆NI8-1 transduction of hippocampal neurons led to electrical coupling, facilitating an increased spontaneous activity. Taken together, these findings strongly support the view that transduced neurons were directly interconnected by membrane fusion.
Fusogenic events may explain the electrophysiological alterations observed in transduced neurons. One plausible interpretation of Rin reduction is an increased membrane surface. Based on the Ohm law, electrical resistance is given by:
where R is the electrical resistance, ρ is the resistivity of the material, L is the length of the material, and A is the cross-sectional area of the material through which the current passes. Because electrical resistance (R) is inversely proportional to the area (A), i.e., to the membrane surface, it is expected to decrease when cells fuse. In presence of a lower Rin, a stronger input would be necessary to trigger an AP in transduced neurons, but, at the same time, after the first AP, fused membranes would facilitate the spreading of AP between cells.
Other electrophysiological alterations cannot be explained directly by a fusogenic effect. For example, the hyperpolarized RMP may be interpreted as a homeostatic response of transduced neurons to increased rates of depolarizing inputs or as a direct consequence of viral envelope fusion with the cell membrane [37]. Therefore, we speculate that other mechanisms capable of modifying host cell biology might take place. For example, alterations in Ca2+ homeostasis have been reported to occur following HSV-1 infection of neuronal cultures [38,39,40]. Indeed, because synaptic activity, in particular excitatory glutamatergic transmission, leads to increases in intracellular calcium concentrations [Ca2+]i, we hypothesized that the hyperactivity of transduced neurons could increase the levels of [Ca2+]i. In Fura-2 experiments, in fact, hippocampal neurons transduced with the J∆NI8-1 vector displayed significantly higher [Ca2+]i compared to controls. In principle, prolonged increases of [Ca2+]i should be cytotoxic but, surprisingly, transduced neurons did not appear to be suffering.
In addition, we found that TTX completely blocked the excessive firing of J∆NI8-1 transduced neurons, indicating sodium dependence of this event. Interestingly, blocking T-type calcium channels mirrored the effect of TTX, suggesting a key role also for T-type calcium channels. Conversely, NBQX, a potent AMPA receptor antagonist, only partially blocked spontaneous spiking of transduced cells, indicating that this event is not dependent on excitatory synaptic activity, and further supporting the view that electrical coupling resulting from fusogenic events is the main driving force for the alterations that we detected.
Altogether, these results demonstrate that fusogenic events might arise after the transduction of neurons with the J∆NI8-1 vector, leading to increased firing frequency, alterations of basic membrane parameters, and increase in basal intracellular calcium levels.
Glycoprotein B plays a key role in membrane fusion processes
As noted, multiple genetic alterations were observed in the J∆NI8-1 vector. Thus, one final question was if any of those could be sufficient to cause the observed phenotypes. As described above, we hypothesize a key role of UL27, the gene coding for gB. Reports in the literature support the view that mutations in gB can enhance cell-cell fusion [41], while others demonstrate the impact of gB on neuronal physiology [37]. For instance, three different strains of replication-competent pseudorabies virus (PRV), which belong to the Herpesviridae family, exhibit distinct effects on neuronal physiology. These strains include the virulent Becker strain (PRV-151), an avirulent Becker strain that does not express gB (PRV-223 gB null), and the attenuated Bartha strain (PRV-152). PRV-151 caused several electrophysiological alterations (high firing frequency, spikelet-like events during hyperpolarization) and the formation of pores between neurons (as demonstrated by both dye and electrical coupling). Similar but delayed effects were observed with the attenuated virus PRV-152. Conversely, no alteration was observed with the gB null mutant PRV-233 [37].
We tested if the mutations that we observed in UL27 were sufficient to produce a fusogenic and hyperexcitable phenotype. Indeed, a newly generated vector batch in which gB NTI was engineered in an otherwise safe J∆NI8 backbone (J∆NI8_A549I; no mutations in other HSV-1 genes) recapitulated the electrophysiological alterations observed in spontaneously mutant batches. Another newly generated vector in which we engineered a gB carrying an NTT mutation, i.e., identical to the above but Alanine converted into Threonine in position 549 (J∆NI8_A549T), did not cause any electrophysiological alteration. Thus, the single most relevant mutation seems to be Alanine converted into Isoleucine at residue 549 (A549I). These results suggest that even subtle mutations in critical regions of gB can dramatically influence viral vector properties, emphasizing the need for a thorough genetic screening during vector development and production.
Implications for gene therapy
Gene therapy has enjoyed success in the past few years. Originally devoted to the treatment of rare monogenic diseases, it seems now to have reached a maturity level for broader approaches to treat diseases affecting millions of people worldwide. Both LV and AAV vectors have been already brought to clinical trials for treating CNS disorders [42,43,44,45]. However, while these vectors are proving effective for some applications, future gene therapies might require the delivery of large genes, complex multigene cassettes, or sophisticated genome editing approaches [46,47,48,49] that greatly exceed their payload capacity. Although several strategies either to miniaturize the expression cassette [50] or to split it in multiple vectors [51] are under development in many laboratories, it remains highly uncertain if these approaches will ultimately result in satisfactory efficacy.
In the past few years, our and other laboratories have made significant advancements in the field of viral vectors based on HSV-1 [23, 49, 52,53,54]. The most attractive feature of these vectors is that they can accommodate very large payloads, amplifying the potential impact of future gene therapy approaches. While last-generation backbones proved very safe in vivo, not expressing any viral protein after direct injection in the brain tissue [22], one prerequisite for applicability to human CNS diseases is the development of quality control measures that ensure the stability of the gene therapy product over multiple rounds of manufacturing.
The possible emergence during propagation of fusogenic mutations in gB that alter neuronal physiology has critical implications for the safety of HSV-1-based vectors, particularly in therapeutic contexts involving the CNS. Syncitial phenotypes have been described in HSV-infected culture, for example, in presence of mutations or truncations in glycoproteins (such as gB, gD, gH/gL, gE, gM, gK) involved in cell fusion [55]. Notably, the emergence of syncytial clones in replication-defective vectors (which, unlike wild-type HSV, have impaired growth) may be explained by the selective pressure for mutations that confer growth advantages. Mutations in the gB gene indeed enhance cell-to-cell spread and this may result in a growth advantage. Moreover, during vector production, there is a tendency to select clones that display strong reporter expression, as these often form more visible plaques. However, such clones may harbor syncytial mutations that become apparent only later. Since plaque selection is typically performed visually, this process is inherently operator-dependent and may not reliably exclude syncytial variants. Moreover, different mutations in the gB gene can confer varying degrees of syncytial activity, making these phenotypes difficult to detect and increasing the likelihood that syncytial clones persist and expand over the following passages.
To mitigate these risks, vector production protocols must incorporate stringent measures to identify and exclude clones harboring these deleterious mutations. High-throughput sequencing and functional assays, as performed in this study, represent essential tools for ensuring vector quality.
This study highlights the crucial need to monitor and control genetic variations in HSV-1–based vectors to ensure their safety for clinical applications. Although rare, DNA recombination may occur during BAC replication or repetitive cycles of cell infection during vector production.
One strategy to mitigate the risk of DNA recombination in specific genes could be to engineer cell lines suitable for viral production to stably express those genes. For instance, the insertion of UL27, providing gB in trans, could greatly reduce the risk of mutations during vector production. Ultimately, future efforts should focus on developing strategies to stabilize the viral genome.

Comments (0)