There is significant heterogeneity between the genotypes and phenotypes of surfactant dysfunction disorders[6]. Our study of surfactant dysfunction-related ILD is, to our knowledge, the first comparative analysis of clinical phenotypes and prognoses across SFTPC, ABCA3, and NKX2-1 genetic mutations. SFTPC mutations predominated in our cohort (50%, 11/22), followed by NKX2-1 (27.3%, 6/22) and ABCA3 (22.7%, 5/22). Although monoallelic ABCA3 mutations are frequently detected in infants with RDS [13, 14], there is insufficient evidence to confirm their pathogenicity. Therefore, only biallelic ABCA3 variants were included herein.
We identified four novel ABCA3 and three NKX2-1 gene variants, thereby expanding the genetic dataset for these genes. In the ABCA3 group, frameshift p.F1295Sfs*51 and deletion p.L438del are classified as pathogenic and likely pathogenic per ACMG criteria, supporting their role in disease. Point mutations p.F255L and p.V337M are classified as uncertain, according to the ACMG criteria's stringent assessment of point mutations. Both of these children with these variants show a classic ILD phenotype, have very low minor allele frequency (MAF), and follow a compound heterozygous pattern, suggesting high pathogenic potential. In the NKX2-1 group, nonsense p.E207* and frameshift p.G266Afs29 are classified as likely pathogenic, while frameshift p.Q373Hfs7 is classified as uncertain by ACMG criteria. All three cases presented a typical NKX2-1-related phenotype, including ILD with hypothyroidism or neurological disorders. Two cases are de novo, and one is inherited from an affected mother, with no MAF data available for these variants in genomic databases, so the variation types and phenotype correlations strongly support their pathogenicity.
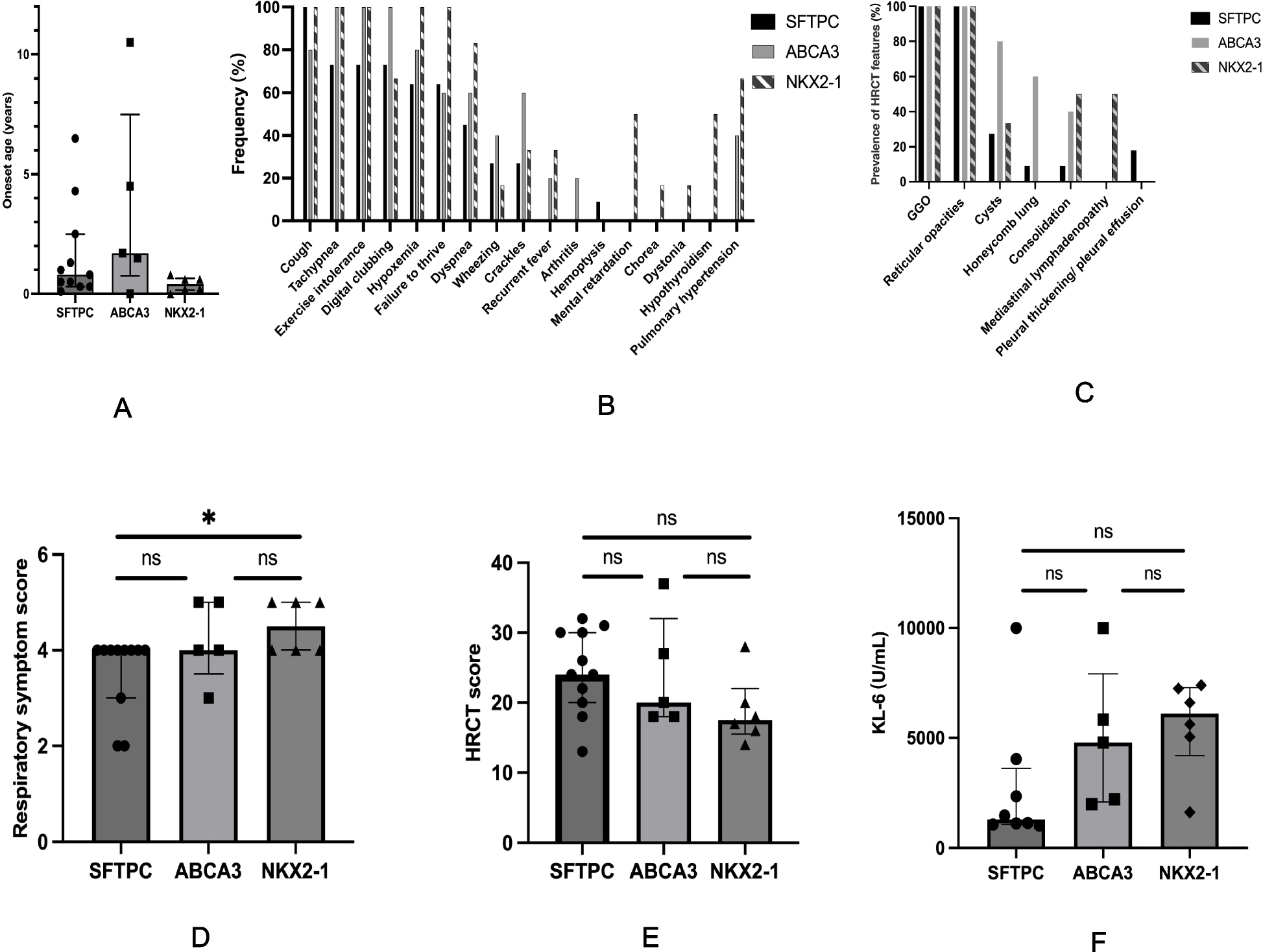
To assess severity and prognosis in surfactant dysfunction-related ILD, we integrated clinical symptoms, HRCT findings, and serum KL-6 levels. KL-6, a biomarker that reflects the degree of alveolar epithelial damage and the severity of ILD [15, 16], demonstrated greater sensitivity in detecting disease changes than HRCT, providing a quantifiable measure of disease progression and treatment response in our study. At initial presentation, the NKX2-1 group had the youngest age of onset and the most severe clinical manifestations, as evidenced by the highest respiratory symptom scores, the highest prevalence of pulmonary hypertension (66.7%), and the highest serum KL-6 levels, followed by the ABCA3 group. Conversely, the SFTPC group showed the mildest clinical manifestations. During follow-up, NKX2-1 variants were associated with the poorest prognosis (50% mortality) and significant deterioration in symptoms and HRCT scores. In contrast, the SFTPC group demonstrated the most favourable outcomes (18.2% mortality), with substantial improvements in symptoms, HRCT findings, and KL-6 levels. Thus, within this cohort, genotype-stratified disease severity followed the gradient NKX2-1 > ABCA3 > SFTPC, whereas prognosis exhibited an inverse relationship. These findings align with previous comparative studies of SFTPC and ABCA3 variants that reported similar clinical, histological and radiological phenotypes but a more severe clinical course in patients with ABCA3 mutation [17]. Notably, no previous studies have directly compared NKX2-1-related ILD with SFTPC- or ABCA3-related ILD; our data identify NKX2-1 as the most severe genotype within our cohort. Although NKX2-1 mutations may manifest as isolated pulmonary involvement, they can also present as part of the classic triad including neurological deficits and hypothyroidism [10]. In our cohort, 50% of NKX2-1-related ILD cases exhibited neurological manifestations or hypothyroidism—features entirely absent in SFTPC and ABCA3 groups—highlighting their potential as genotype-specific diagnostic markers.
Interestingly, autoimmune processes were identified in 27.3% of patients across all three genotypes, with elevated autoantibodies including RF, CCP, ANA, and ANCA. Notably, one patient in the ABCA3 group (P16) exhibited rheumatoid arthritis, characterized by arthritis and elevated levels of RF, CCP, and ANA. Additionally, the father of one patient with an SFTPC mutation (P2) also presented with ILD and rheumatoid arthritis. Consistent with our findings, a prior study observed the co-occurrence of rheumatoid arthritis and ILD in an adult with an SFTPC mutation [18]. To rule out the potential influence of co-existing genes that could lead to autoimmune manifestations, we exhaustively analyzed WES data for genes such as COPA, STING1/TMEM173, LRBA, CTLA4, STAT1, STAT3, and others linked to autoimmune or immunodeficiency conditions, identifying no pathogenic mutations. These findings suggest a potential role for autoimmunity in the pathogenesis of surfactant dysfunction disorders.
In our cohort, surfactant dysfunction–related lung disease encompassed not only ILD but also DAH. Notably, DAH can be clinically subtle: one patient with an SFTPC variant presented without hemoptysis or anemia and was diagnosed solely on the basis of extensive GGO on HRCT and abundant hemosiderin-laden macrophages in BALF, highlighting the critical diagnostic role of bronchoscopy. The co-occurrence of ILD and DAH is rare and has been reported in diseases with autoimmunity, including ANCA-associated vasculitis [19], COPA syndrome [20], STING-associated vasculopathy with onset in infancy (SAVI) [21], et al. Consistent with these observations, all three DAH cases in our series exhibited elevated autoantibody levels, suggesting that autoimmunity may play a contributory role in the development of DAH within surfactant dysfunction disorders. The presence of DAH may exacerbate disease severity, complicate treatment, and even induce mortality. Therefore, timely identification and aggressive management of DAH are essential.
Surfactant dysfunction-related ILD lacks a standardized treatment protocol. CS and HCQ have shown effectiveness in some cases of SFTPC, ABCA3 and NKX2-1-related ILD [13, 22,23,24,25,26,27,28,29,30]. For SFTPC dysfunction, the therapeutic efficacy of HCQ is attributed not only to its anti-inflammatory properties but also to its potential to influence the cellular processing of surfactant protein C precursors [23]. For ABCA3 dysfunction, HCQ targets lysosome-related lamellar bodies, which are integral to ABCA3's normal function in surfactant lipid transport. As a weak base, HCQ accumulates in acidic compartments, potentially modulating vesicular pH and promoting ABCA3 protein processing [29]. However, the therapeutic efficacy of HCQ in ABCA3-related ILD appears to be variant-dependent. An ABCA3 + vesicle volume exceeding 60% of the wild-type volume has been associated with responsiveness to HCQ [29]. The efficacy of CS and HCQ across different genetic mutations has not been systematically analyzed. In our study, 77.5% of patients received combination therapy with CS and HCQ. The SFTPC group exhibited the most favorable response, with 87.5% showing improvement in both symptoms and HRCT findings, and 83.3% achieving a > 20% reduction in KL-6 levels. In contrast, the NKX2-1 group demonstrated the poorest outcomes, with only 40% showing symptom improvement and 25% exhibiting HRCT and KL-6 improvement. Mortality rates varied significantly: 0% in the SFTPC group, 25% in the ABCA3 group, and 60% in the NKX2-1 group. These results suggest that CS + HCQ combination therapy is the most effective for ILD in the SFTPC group, whereas alternative treatment strategies may be required for NKX2-1-associated ILD.
The optimal treatment duration for HCQ in surfactant dysfunction-related ILD requires further investigation. While potential adverse effects may occur with prolonged HCQ use, a recent study involving 397 pediatric patients with chronic conditions demonstrated an excellent safety profile, with only one reported case of cutaneous adverse reaction observed over a mean treatment duration of 29.6 months [31]. In our study, the longest treatment duration for HCQ was 69 months, with no HCQ-related adverse effects observed in any patient. Within the SFTPC group, one patient who discontinued CS and continued HCQ as maintenance therapy has maintained stable clinical manifestations. However, one of two patients who discontinued both CS and HCQ experienced relapse. In the ABCA3 group, two patients maintained stable disease after discontinuing CS and continuing HCQ monotherapy, whereas one of them experienced disease exacerbation following the cessation of HCQ, with improvement noted again after reintroducing HCQ. These findings suggest that HCQ may play a more critical role in the maintenance treatment of SFTPC- and ABCA3-related ILD, indicating a potential need for long-term use.
Regarding other pharmacological treatments, previous studies have suggested potential benefits of azithromycin in ABCA3-related ILD [32, 33]. In our study, one patient in the ABCA3 group died despite azithromycin monotherapy, while one patient in the NKX2-1 group maintained clinical stability when azithromycin was used as maintenance therapy following initial treatment with CS and HCQ. Due to the limited number of cases, the therapeutic efficacy of azithromycin in surfactant dysfunction-related ILD requires further investigation. Notably, in a case of SFTPC-related ILD complicated by DAH, combined CS and cyclophosphamide therapy resulted in significant clinical improvement, highlighting the potential utility of immunosuppressive regimens in patients with DAH. The efficacy of antifibrotic therapy also remains unclear. One ABCA3-related ILD patient receiving pirfenidone monotherapy for five months (after discontinuation of CS and HCQ) exhibited neither symptomatic improvement nor reduction in persistently elevated KL-6 levels. Prospective studies are needed to determine whether antifibrotic agents, either as monotherapy or in combination regimens, confer clinical benefit in surfactant dysfunction-related ILD. Recent advances highlight promising targeted pharmacological approaches. High-content screening has identified cyclosporin A as a molecular corrector for specific ABCA3 variants [34], potentially offering an alternative for hydroxychloroquine-refractory cases. Furthermore, emerging gene therapy approaches may provide future opportunities for personalized treatment strategies [6].
There are several limitations to our study. First, the cohort was derived exclusively from the Department of Respiratory Medicine and did not include patients from neonatal units. Consequently, severe presentations of surfactant dysfunction disorders that lead to fatal outcomes during the neonatal period were not represented in this analysis. Second, due to the rarity of the disease, the number of cases included in each group was relatively small. Third, we did not perform functional assays to ascertain the effects of the novel genetic variants on protein function. Future studies should address these limitations to provide a more comprehensive understanding of the disease spectrum and the functional consequences of identified genetic variants.



Comments (0)