Cell culture and treatment conditions
NCI-H1581 human lung cancer cells (CRL-5878, ATCC) were maintained in RPMI-1640 medium (Gibco, 11875093) enriched with 10% heat-inactivated FBS (Gibco, 16000044), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco, 15140122). Cells were incubated at 37 °C under 5% CO₂ humidification. For experimental procedures, cells were seeded into 6-well plates (Corning, 3516) at 5 × 104 cells/cm2 or white 96-well plates (Corning, 3917) at 5 × 104 cells/well for luminescence assays, and permitted to adhere for 24 h. PYY1-36 (Tocris Bioscience, 3858) was reconstituted in sterile PBS (Gibco, 10010023) to generate a 1 mM stock, then diluted to 0 (vehicle, PBS), 2.5, 5, 10, 25, 50, 100, or 200 nM in fresh medium. Treatments spanned 48 h. Lentiviral transduction involved exposing cells to RBM43-targeting shRNA (Sigma-Aldrich, TRCN0000421352) or scrambled control (Sigma-Aldrich, SHC002) at MOI 5 for 24 h, followed by 72-h puromycin selection (2 μg/mL; Sigma-Aldrich, P9620). Each condition included three technical replicates, with six independent experiments (n = 6), unless otherwise stated.
Lactate dehydrogenase (LDH) release assay
Cytotoxicity was quantified using the CytoTox 96 Assay (Promega, G1780). NCI-H1581 cells in 96-well plates were treated with 0 (vehicle, PBS), 2.5, 5, 10, 25, 50, 100, or 200 nM PYY1-36 for 48 h. Post-treatment, 50 μL supernatant was added to a 96-well plate (Corning, 3599) with 50 μL substrate. After 30 min at 22 °C in the dark, 50 μL stop solution was added. Absorbance at 490 nm was read on a SpectraMax M5 (Molecular Devices). LDH release (%) was calculated as: [(Experimental OD – Background OD)/(Maximum LDH OD – Background OD)] × 100. Maximum LDH was determined using 1% Triton X-100 (Sigma-Aldrich, T8787) for cell lysis.. Data represent mean ± SEM from six independent experiments (n = 6) with three technical replicates.
Cell viability assay
Cell viability was evaluated by the MTT assay. NCI-H1581 cells (5 × 103 cells/well) were seeded in 96-well plates, and treated with PYY1-36 (0–200 nM) for 48 h. MTT (0.5 mg/mL) was added for 4 h. Absorbance at 570 nm was measured using a SpectraMax M5 reader. Viability was expressed as a percentage of the vehicle control. Experiments were done in triplicate with six repeats (n = 6).
ROS-Glo H₂O₂ assay for hydrogen peroxide quantification
NCI-H1581 cells were seeded at 5 × 104 cells/well in white 96-well plates (Corning, 3917) in RPMI-1640 medium (Gibco, 11,875,093) supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, and incubated at 37 °C with 5% CO₂ for 24 h to adhere. Cells were treated with 0 (vehicle, PBS), 50, or 100 nM PYY1-36 for 48 h, with controls: positive (100 μM H₂O₂, Sigma-Aldrich, H1009, for 1 h) or antioxidant (5 mM N-acetylcysteine, NAC, Sigma-Aldrich, A7250, for 1 h before 100 nM PYY1-36). The ROS-Glo H₂O₂ Assay (Promega, G8820) was performed per the manufacturer’s protocol, as an alternative to flow cytometry due to poor-quality MitoSOX Red data. Briefly, H₂O₂ Substrate Solution (25 μM) was added to each well for the final 6 h of treatment. After 6 h, 100 μL of ROS-Glo Detection Solution (containing luciferase and D-cysteine) was added, and plates were incubated for 20 min at room temperature to generate a luminescent signal proportional to H₂O₂ levels. Luminescence was measured using a GloMax Navigator microplate luminometer (Promega). Total protein content was quantified in parallel wells using the BCA Protein Assay (Pierce, 23,225), and luminescence values were normalized to protein content (relative luminescence units, RLU/μg protein, vehicle set as 1.0). Each condition included three technical replicates, with six independent experiments (n = 6).
Quantification of 8-Hydroxy-2’-Deoxyguanosine (8-OHdG)
NCI-H1581 cells in 6-well plates were treated with 0 (vehicle, PBS), 50, or 100 nM PYY1-36 for 48 h. DNA was purified using the DNeasy Kit (Qiagen, 69,504), quantified, and hydrolyzed. Hydrolyzed DNA (50 μL at 1 μg/μL) was applied to 8-OHdG ELISA plates (Cell BioLabs, STA-334), incubated with anti-8-OHdG antibody (1:100, 1 h, 22 °C), then HRP-conjugated secondary antibody (1:2000, 1 h, 22 °C). Colorimetric development with TMB substrate was halted using 2 N H₂SO₄, and absorbance at 450 nm was recorded (SpectraMax i3x). Values were standardized to total protein (pg/g protein). Data represent mean ± SEM from six independent experiments (n = 6) with three technical replicates.
ATP measurement
NCI-H1581 cells in white 96-well plates were treated with 0 (vehicle, PBS), 50, or 100 nM PYY1-36 for 48 h. ATP levels were determined via the CellTiter-Glo® 2.0 Assay (Promega, G9242). Cells were lysed with 100 μL/well lysis buffer, and 50 μL lysate was blended with the reagent in white 96-well plates (Corning, 3917). Luminescence was captured after 10 min (GloMax® Navigator). ATP concentrations (nmol/mg protein) were normalized to total protein (BCA Assay, Pierce™ 23,225). Data represent mean ± SEM from six independent experiments (n = 6) with three technical replicates.
Oxygen consumption rate (OCR)
NCI-H1581 cells (2 × 104/well) in XF24 plates (Agilent, 100,777–004) were treated with 0 (vehicle, PBS), 50, or 100 nM PYY1-36 for 48 h. OCR was analyzed using the Seahorse XF Mito Stress Kit. Cells (2 × 104/well) in XF24 plates were treated with PYY1-36 for 48 h. Before analysis, cells were equilibrated for 1 h in XF Base Medium with 10 mM glucose, 1 mM pyruvate, and 2 mM glutamine (pH 7.4). OCR was measured under basal conditions and after adding 1 μM oligomycin, 2 μM FCCP, and 0.5 μM rotenone/antimycin A. Basal and maximal OCR were expressed as pmol/min/104 cells. Data represent mean ± SEM from six independent experiments (n = 6) with three technical replicates.
Complex I activity
NCI-H1581 cells in 6-well plates were treated with 0 (vehicle, PBS), 50, or 100 nM PYY1-36 for 48 h. NADH dehydrogenase activity was determined spectrophotometrically. Mitochondria isolated (Abcam kit ab110168) were lysed in 25 mM KH₂PO₄ buffer (pH 7.2) containing 1% n-dodecyl-β-D-maltoside (Sigma-Aldrich, D4641). Reactions commenced with 100 μM NADH (Sigma-Aldrich, N8129) and 50 μM coenzyme Q1 (Sigma-Aldrich, C7956). Absorbance at 340 nm was monitored for 10 min (SpectraMax M5). Activity (nmol/min/mg protein) was computed using ε = 6.22 mM⁻1·cm⁻1. Data represent mean ± SEM from six independent experiments (n = 6) with three technical replicates.
MitoTracker red fluorescence assay
NCI-H1581 cells in 6-well plates were treated with 0 (vehicle, PBS) or 100 nM PYY1-36 for 48 h. Mitochondrial mass was quantified using MitoTracker Red CMXRos (Thermo Fisher, M7512). Cells were stained with 100 nM MitoTracker Red in RPMI-1640 for 30 min at 37 °C, washed twice with PBS, and resuspended in PBS. Staining was validated using unstained cells and positive control (50 μM CCCP, Sigma-Aldrich, C2759, for 1 h to depolarize mitochondria). Fluorescence was measured by flow cytometry on a BD FACSCelesta (581/644 nm excitation/emission). A minimum of 10,000 events per sample were analyzed, with gating to exclude debris and doublets using forward and side scatter. PMT voltages and gains were optimized to ensure signals were within the linear range, with compensation applied as needed. Data were presented as histograms with scale bars indicating fluorescence intensity (x-axis, log scale) and cell count (y-axis). Mean fluorescence intensity was quantified using FlowJo v10.8.1, normalized to vehicle (set as 1.0). Data represent mean ± SEM from six independent experiments (n = 6) with three technical replicates.
Quantification of mitochondrial DNA (mtDNA) to nuclear DNA (nDNA) ratio
NCI-H1581 cells in 6-well plates were treated with 0 (vehicle, PBS) or 100 nM PYY1-36 for 48 h. mtDNA/nDNA ratios were measured by qPCR. DNA extracted with DNeasy Kit (Qiagen) was amplified using SYBR Green (Applied Biosystems) on StepOnePlus. ND2 (mtDNA) and B2M (nDNA; Origene) primers were used. Cycling: 95 °C (10 min), 40 cycles of 95 °C (15 s), 60 °C (1 min). Ratios were calculated using the 2−ΔΔCt method, normalized to vehicle (set as 1.0). Data represent mean ± SEM from six independent experiments (n = 6) with three technical replicates.
RNA extraction and quantitative PCR (qPCR)
NCI-H1581 cells in 6-well plates were treated with 0 (vehicle, PBS) or 100 nM PYY1-36 for 48 h, or transduced with RBM43 shRNA and treated as described. RNA was isolated using the RNeasy Mini Kit (Qiagen, #74,104), with purity/concentration checked by NanoDrop 2000 (Thermo Fisher Scientific). cDNA was synthesized from 1 μg RNA using the High-Capacity Reverse Transcription Kit (Applied Biosystems, #4,368,813). qPCR was performed on a StepOnePlus system (Applied Biosystems) with SYBR Green Master Mix (Applied Biosystems, #4,367,658). Primer sequences: TFAM: Forward 5′-ATGGCGTTTCTCCGAAGCAT-3′, Reverse 5′-TCCGCCCTATAAGCATCTTGA-3′; NRF1: Forward 5′-AGGAACACGGAGTGACCCAA-3′, Reverse 5′-TATGCTCGGTGTAAGTAGCCA-3′ Cycling: 95 °C (10 min), 40 cycles of 95 °C (15 s) and 60 °C (1 min). mRNA levels were normalized to GAPDH (2−ΔΔCt method, expressed relative to vehicle (set as 1.0). Data represent mean ± SEM from six independent experiments (n = 6) with three technical replicates.
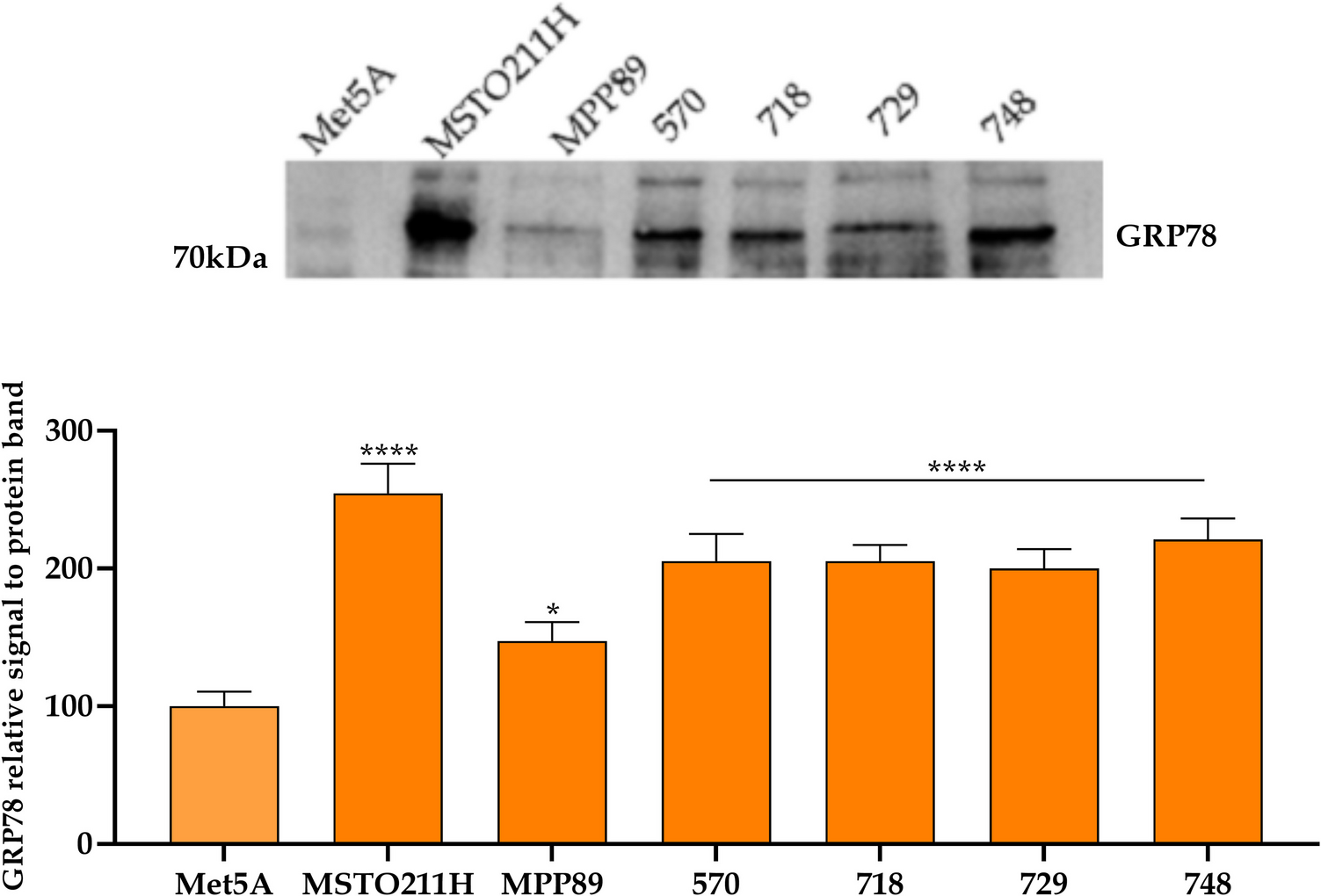
Western blot analysis
NCI-H1581 cells in 6-well plates were treated with 0 (vehicle, PBS) or 100 nM PYY1-36 for 48 h, or transduced with RBM43 shRNA and treated as described. Proteins were extracted with RIPA buffer (Thermo Fisher, 89,900) containing protease inhibitors (Roche, 11,836,170,001). Samples (20 μg/lane) were resolved on 10% SDS-PAGE gels (Bio-Rad, 4,561,033) and transferred to PVDF membranes (Millipore). Membranes were blocked in 5% non-fat milk/TBST for 1 h at RT, then probed overnight at 4 °C with primary antibodies: anti-ND1 (Proteintech, 19,703–1-AP; 1:1000), anti-SDHB (Abcam, ab14714; 1:1000), anti-UQCRC2 (Proteintech, 14,742–1-AP; 1:1000), anti-MTCOX2 (Abcam, ab198286; 1:1000), anti-ATP5 A1 (Cell Signaling, 18,023; 1:1000), anti-NRF1 (Abcam, ab34682; 1:1000), anti-TFAM (Cell Signaling, 7495; 1:1000), anti-PGC-1α (Abcam, ab191838; 1:1000), anti-RBM43 (Novus Biologicals, NBP1-86,798; 1:1000), and anti-β-actin (Abcam, ab8227; 1:5000). After TBST washes, membranes were incubated with HRP-secondary antibodies (Cell Signaling, 7074; 1:3000) for 1 h at RT. ECL detection (GE Healthcare, RPN2132) and ImageJ quantification followed, with bands normalized to β-actin and expressed relative to vehicle (1.0). Data are mean ± SEM from six experiments (n = 6) with three replicates.
ROS-Glo H₂O₂ assay for hydrogen peroxide quantification
Hydrogen peroxide (H₂O₂) levels were quantified using the ROS-Glo H₂O₂ Assay (Promega, G8820). NCI-H1581 cells were seeded at 5 × 104 cells/well in white 96-well plates (Corning, 3917) and treated for 48 h with vehicle (PBS), PYY1-36 (50 or 100 nM), or controls: positive (vehicle with 100 μM H₂O₂ for 1 h) or antioxidant (5 mM NAC for 1 h before 100 nM PYY1-36). H₂O₂ Substrate Solution (25 μM) was added for the final 6 h of treatment, followed by 100 μL ROS-Glo Detection Solution for 20 min at room temperature. Luminescence was measured on a GloMax Navigator luminometer (Promega). Luminescence was normalized to total protein content (BCA assay, Pierce, 23,225) and expressed as relative luminescence units (RLU) per μg protein. Each condition included three technical replicates, with six independent experiments (n = 6).
Statistical analysis
Results are mean ± SEM from six experiments (n = 6). Statistical significance was determined by one-way ANOVA with Tukey’s test using GraphPad Prism 9.0; P < 0.05 was considered significant.

Comments (0)