
Remember me



The two questionnaires (OpERA and PEER-Regulatory) were completed by the Heads of Registration Department of all seven EAC-MRH member countries. In addition, 25 manufacturers were determined to be eligible for this study. There were 11 non-responders in this group, leading to a 56% response rate (14 responders), all of whom completed the PEER-Industry questionnaire. These study participants were distributed into three categories: generics (foreign), that is applicants who manufacture generic medicines outside the EAC region; generics (local), that is applicants who manufacture generic medicines within the EAC region; and innovators, that is applicants who submitted applications for registration of innovator medicines. During the period of study, there were no local innovators. From the results of this study, it was noted that the regulatory review processes of these authorities vary and require further alignment. A point in case is the clock-stop time, that is, the period during which regulatory review stops while an applicant answers regulatory authority questions regarding an application. Clock-stop times vary from authority to authority, making it difficult to compare the actual review timelines against the target timelines. Other differences in regulatory review processes include those of target timelines for review models as well as disparities in target timeline for start and finish of expert committee reviews. The key recommendations from this study were to invest in regulatory systems strengthening, streamline country processes, and minimise the differences that exist within NRAs as these interventions will improve patients’ access to safe, high-quality, and effective medical products, especially during the operationalisation of the African Medicines Agency (AMA).
This study also proposed a very important recommendation, which is the need to review the operating model of the EAC-MRH programme so as to identify areas of improvement regarding the effectiveness and efficiency of the initiative. All seven NRAs in the region and 14 out of the 25 pharmaceutical companies who had submitted applications through the EAC-MRH process from 2015 to 2022 participated in this study. This study resulted in the identification of the successes and challenges of the EAC-MRH after 10 years of implementation regarding regulators, the pharmaceutical industry, and patients and proposed measures that can improve the effectiveness and efficiency of the work-sharing initiative.
3.1 Successes of the EAC-MRHThis initiative has developed harmonised technical requirements and guidelines for the regulation of medical products together with a compendium of established common technical documents (CTD) to provide harmonised medicines registration procedures [2]. Median timelines for joint reviews from submission of application to when a decision is made have decreased [3], and the timelines for registration of medical products have also been reduced by almost half [6]. It was affirmed that this initiative has improved regulatory capacity for NRAs in the region, especially as it has provided a platform for information sharing and learning from best practices [5]. A key benefit for pharmaceutical companies using the work-sharing initiative to apply for marketing authorisation was the reduced burden associated with the need to prepare only one application (modules 2–5) for submission to many countries and eventual access to many markets simultaneously [7]. Applicant time and resources are also conserved by the need to prepare only one response package for a consolidated list of queries from many countries. Shorter timelines for approval of applications through the EAC process as compared to some country processes were also identified as a key success factor for the initiative. The benefit of this process for patients is that the harmonised and working efforts have resulted in quicker access to quality-assured medicines and increased availability [7].
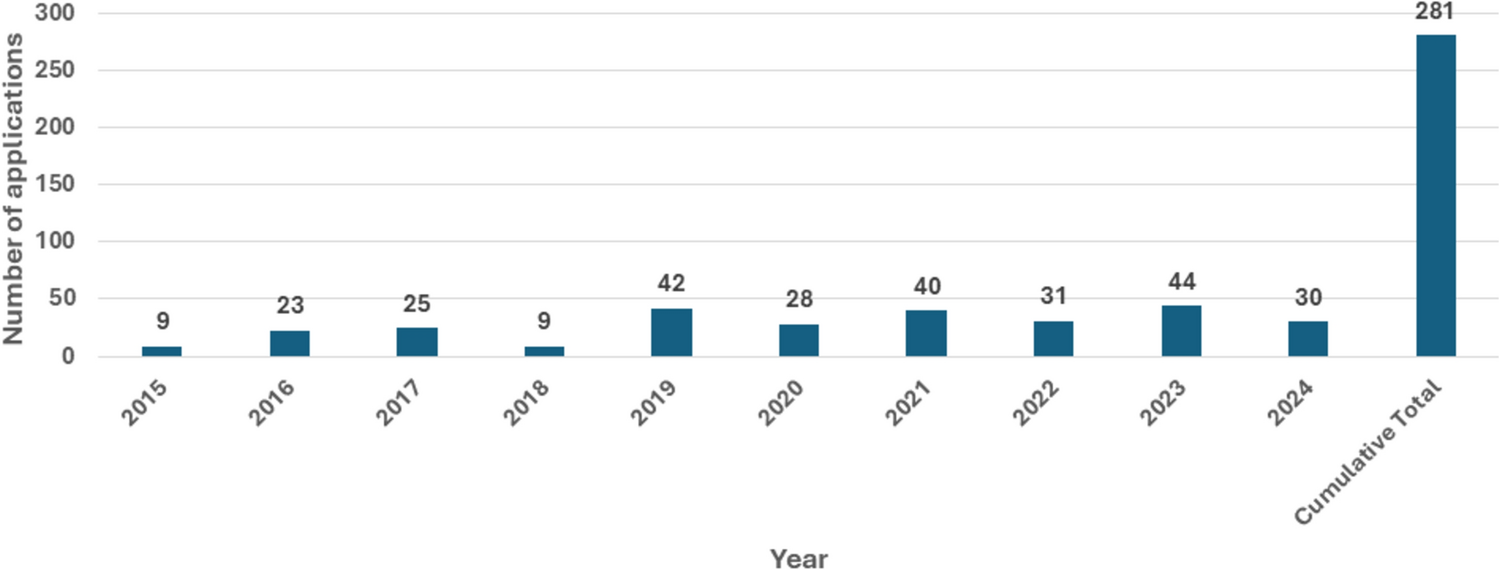
Positively, the number of applications received for joint reviews increased from nine in 2015 to 44 in 2023 (Fig. 1). Review timelines have significantly reduced from 2015 to 2023, with a 53% decline in the median time at the NRA level (Fig. 2). The median time represents a positive regional joint assessment including manufacturer and regional assessment time for the cohort of applications that received a positive regional assessment in that calendar year.
Fig. 1
Cumulative trend of product applications in the EAC-MRH (2015 to Feb 2024). EAC-MRH East African Community Medicines Regulatory Harmonization
Fig. 2
Median timelines for medicines per year recommended for registration by EAC-MRH (2015–2023). EAC-MRH East African Community Medicines Regulatory Harmonization
3.2 The Proposed New Improved Review Model for EAC-MRHTo ensure clarity, the results will be presented in three parts: part 1—a proposed improved model for the EAC NRAs; part 2—proposed improvements to the current operating model of the EAC-MRH initiative; and part 3—a proposed new improved model for the EAC-MRH initiative.
3.3 Part 1: Proposed Improved Model to the EAC NRAsThe regulatory review systems of the NRAs in the EAC region need to be strengthened so as to improve the effectiveness and efficiency of the EAC-MRH work-sharing initiative and eventually the AMA when it is operational. These are some proposals for implementation by the NRAs to improve their regulatory review systems.
3.3.1 Legal FrameworksOne of the key challenges faced by NRAs that stimulated the establishment of regulatory harmonisation was the fragmented legal frameworks of countries in Africa. The NRAs in the EAC region are called upon to domesticate the African Union (AU) Model Law on Medical Products Regulation [12]. The AU model was endorsed by the AU Heads of State and Governments in 2016. “The purpose of this law is to establish an effective and efficient system of medical products regulation and control and ensure that such products meet required standards of safety, efficacy and quality” [13]. This is a non-prescriptive piece of legislation expected to be domesticated and implemented by all the AU member states and regional economic communities (RECs) with the objectives of increasing NRA partnerships, harmonising regulatory systems, and eventually providing favourable conditions for increased development of medicines and medical technologies [4] (Fig. 3). It describes the essential features and requirements that must be included in the regulatory system and offers African nations a template for harmonising their regulatory systems [6].
Fig. 3
Source: AUDA-NEPAD website. AU African Union, AUDA-NEPAD African Union Development Agency – New Partnership for Africa’s Development
The AU Model Law on Medical Products Regulation.
The AU Model Law is also intended to assist countries in incorporating the ability to charge, collect, and utilise fees for services carried out during the examination or enactment of their laws. Collection of fees for the harmonisation initiative has not been implemented yet due to a delay in the development and approval of the sustainability plan for this initiative. The development of the sustainability plan started in 2018 and, due to some political hurdles, was only approved in 2024 by the Sectoral Council. There are ongoing preparatory efforts toward the implementation of fees. Domestication of the law will ensure that the authorities in the region have comprehensive laws for regulation of medical products and eventually facilitate the harmonisation process of the EAC-MRH initiative. Domesticating the AU Model Law will also address the obstacle regarding the delay of national decisions after a regional recommendation due to national laws on reliance. The harmonisation initiative will not have any value if regional recommendations do not translate into countries granting marketing authorisation to the recommended medical products. Legislation that specifically allows for national authorisations based on reliance on documents/recommendations from trusted reference authorities or initiatives (like the EAC-MRH) is key [4].
According to Ncube and colleagues [4], only four NRAs (ABREMA, Burundi; KPPB, Kenya; TMDA, Tanzania Mainland; and ZFDA, Tanzania Zanzibar) out of the seven in the region have domesticated the AU Model Law. Several reasons have been highlighted on why some countries have not domesticated the law: “Lack of human and financial resources, competing priorities at the national level, overlapping roles of government institutions, and the process of amending/repealing laws being slow and lengthy” [4].
3.3.2 Benefit-Risk AssessmentFor NRAs to rely on each other or harmonise medicine regulation, there is a need for them to use standardised templates that will enable quality decision-making processes and transparency. Although regulatory authorities may receive applications that have the same information from manufacturing authorities, they may make different decisions, as most of them use checklists for their review. There is now a growing interest from regulatory authorities to use a more structured approach for decision making and transparency. A consistent and transparent benefit-risk assessment decision is based on a structured flow of information and the systematic approach to assessing the benefits and risks, which is well documented and communicated to relevant stakeholders for accountability purposes [14, 15]. It is important that the key players such as patients, medical practitioners, and regulators identify with the regulatory decisions being made. Nowadays, to improve transparency and accountability and to be in line with good review practices, regulatory authorities are facing a great deal of pressure to implement a systematic and structured approach in making regulatory decisions on the benefit-risk assessments of medical products [16]. Regulators are expected to make a balanced judgement regarding the benefits and risks of a new medical product that is being brought to the market and communicate this judgement to the public as one of the measures to enhance regulatory effectiveness [15].
How do authorities in the EAC region document and communicate the benefits and risks of a medical product? The benefit-risk assessment process is not yet implemented in this region. The Centre for Innovation in Regulatory Science (CIRS) has developed an eight-step (Fig. 4) Universal Methodology for Benefit-Risk Assessment (UMBRA) that can be used by NRAs in the EAC region to document benefit-risk assessments in a structured and systematic way [17].
Fig. 4
UMBRA benefit-risk framework. Reprinted [15]. UMBRA Universal Methodology for Benefit-Risk Assessment
3.3.3 Building NRA CapacityOnly one NRA in this study reviews applications on NASs; however, it is important to empower the NRAs to be able to review applications for novel medicines, a capacity that may become increasingly relevant during emergency situations. NRAs should also invest more in human resources to be able to respond in a timely manner to high demands for their services.
3.3.4 Develop and implement a robust regulatory information management systemThe AMRH programme has recommended a model regulatory information management system (RIMS) for countries that do not have information management systems for use by the NRA. A robust RIMS should be developed by each NRA in the region to provide online and real-time medicine regulation information and support workflow management in the authority, as this will assist in the management of data during the review process. The RIMS should contain metric tools that countries can use to track applications and capture data on key milestones throughout the registration process. NRAs should also implement the electronic common technical document (e-CTD), which is the digitalised way to accelerate assessment reviews. The RIMS should be interoperable and can be integrated with the RIMS of other NRAs in the region and also linked to the regional EAC-MRH system and eventually the continental RIMS when AMA becomes operational (Fig. 5). The Regulatory Information Sharing Portal (RISP) being developed by the AMRH Programme in African Union Development Agency – New Partnership for Africa’s Development (AUDA-NEPAD) should be able to extract key regulatory information from national RIMS and the regional EAC-MRH system to share at the continental level (Fig. 5). Countries are called upon to develop their websites and make publicly available all products recommended through the MRH process and which are granted marketing authorisation in the country. To ensure effective implementation of RIMS by NRAs, the AMRH Information Management System Technical Committee (IMS TC) has developed a digitalisation strategy for RIMS in Africa to guide countries as they develop their robust information management systems (Fig. 6). It is important for all the EAC NRAs to customize this strategy and use it to develop their systems to enable interoperability of systems in the region.
Fig. 5
Regulatory information sharing portal (RISP) links to national regulatory authorities (NRAs), regional economic communities (RECs), and African Medicines Regulatory Harmonisation (AMRH)/African Medicines Agency (AMA)
Fig. 6
Six strategic priorities for implementing regulatory information management systems (RIMS) in Africa
3.3.5 Implementation of Target Timelines by NRAsAll member states should use 90 calendar days as the target timeline to register products after an application has been received by the NRA after the regional recommendation. A joint recommendation should be made for the application and a joint good manufacturing practice (GMP) inspection conducted or GMP decision made (GMP compliance) before the clock starts. Currently, a great deal of time is often lost when an applicant delays submitting their application to the NRA of interest after the regional recommendation is made, and applicants should be given a target timeline for submitting these applications. Such target timelines have been implemented in the West Africa and Southern Africa work-sharing programme, where a maximum of 2 years and 1 year, respectively, is given to applicants to submit their application to the country of interest after the regional recommendation, after which the application must be re-submitted for review again at the regional level. Part of the reasons for this delay in granting marketing authorisation is that some countries may start their review process again after they receive a regional recommendation. Some countries have different timelines when the scientific committees, meet which might not align with the time when the regional recommendations are made. Using the SADC model, 1 year is being proposed to the EAC-MRH. Countries should track the progress of each application from receipt of application to marketing authorisation.
3.3.6 Implementation of RelianceIn the EAC region, Tanzania and Rwanda have attained World Health Organization (WHO) Global Benchmarking Tool maturity level 3 (WHO GBT ML3). It is clear that not all countries can attain the ML3 status in the near future, and it is therefore imperative for the NRAs to rely on the more resourced WHO-listed regulatory authorities and the EAC-MRH work-sharing initiative. In a study to evaluate the impact of reliance in an NRA and how it improves patient access to medical products, Danks et al. [18] demonstrated how, through the use of an abridged review for NASs and generics, it reduced regulatory review times from 179 days for a full review to 91 days for an abridged review [19]. Countries in the region are therefore called on to domesticate continental guidelines developed by the AUDA-NEPAD Technical Committees to enhance the harmonisation process.
3.4 Part 2: Proposed Improvement to the Current Work-Sharing Operating Model of the EAC-MRH InitiativeThe current work-sharing model for the EAC-MRH initiative is shown in Fig. 7. Initially, the EAC-MRH lead authority receives applications for joint review only when the applicant has paid the application fees to two or more countries in the region. A framework should be developed to enable a centralised regional submission and review prior to submission to the individual countries of interest for registration. Consideration should be given to using three routes/procedures for the approval of medical products in the region; that is, a fully centralised procedure, a decentralised procedure, and a national procedure. In each of these approaches, full, abridged, or verification procedures will still be conducted in the revised model. These new procedures will not replace the full, abridged, and verification procedures, but will rather be new legal frameworks under which these three approaches to an assessment can be conducted. In order to enable the creation of a completely centralised approach similar to that which is implemented in the European Union (EU), it would be necessary for the region to pursue the creation of a regional legally enforceable framework (Fig. 8). Regardless of legislative maturity or capacity, the adoption of the centralised procedure might be made mandatory for some essential medical products to provide appropriate access in all member states. Another advantage of a centralised process is the use of local specialists in central safety monitoring and the assessment of complex items (Fig. 8).
Fig. 7
Current review process map and milestones for East African Community (EAC) joint assessment procedure. Reprinted [7]. GMP good manufacturing practice, MA marketing authorisation, NMRA national medicines regulatory authority
Fig. 8
Proposed new East African Community Medicines Regulatory Harmonization (EAC-MRH) programme centralised procedure. EAC East African Community, GMP good manufacturing practice, MA marketing authorisation, NRA national regulatory authority
3.4.1 GMP InspectionsThe two routes for applicants for EAC GMP inspection are the country process or the joint inspection process. Joint inspections entail inspectors from the participating countries inspecting a manufacturing site together. Some delays with GMP are caused when applicants have not paid joint GMP inspection fees but pay the country fees, after which the country initiates the GMP process. Ideally, however, products that are jointly reviewed should be jointly inspected. There are cases where manufacturers or applicants do not submit an application for GMP because a previous GMP audit is still valid or compliant and they have been inspected by two or three well-resourced NRAs such as the TMDA, KPPB, or NDA. In such cases, the GMP Technical Working Group (TWG) will review the reports of these NRAs that have inspected the site and consolidate the report and then make a recommendation. The GMP lead NRA for EAC-MRH is the NDA and should continue to be pragmatic in combining joint GMP and country processes. It is important to combine regional GMP and national decisions. A document review should be encouraged, especially as the resources are minimal and the standard operating procedures (SOPs) need to be drafted by the technical team.
3.4.2 Reliance and Review ModelReliance mechanisms should be implemented both at the regional and national levels. “The RECs should continue to support and advocate the strengthening of the capacity of their member states using the WHO GBT assessments and other tools such as Optimising Efficiencies in Regulatory Agencies (OpERA) and the Quality of Decision-Making Orientation Scheme (QoDoS) to facilitate inter-country and inter-REC reliance including unilateral and mutual recognition.” Inter-REC reliance should be promoted among the RECs, and if one REC has recommended a product for registration, the other RECs implementing the MRH programme should also rely on this decision, using an abridged or verification review process [19, 20].
A WHO Technical Report in 2021, suggests the use of a standard pathway for marketing authorisation “which entails independent decision making and complete review of the application by NRAs”. This might involve using the CTD format of dossier that has a long registration timeline. The work-sharing pathway allows for possible concurrent or parallel decision making such as REC joint assessments. In addition, it would then be possible to observe and rely on the review in EU-Medicines for all (EU-M4all) or Swissmedic Marketing Authorisation for Global Health Products [21].
Reliance pathways entail the NRA decision being dependent on those made by trusted regulators, a unilateral or mutual recognition pathway, risk-based pathways, abridged review, verification of sameness review, WHO collaborative registration procedure (CRP), or regional reliance pathways (ZAZIBONA, EAC, ECOWAS).
It is critical to ensure that the MRH initiative has a legal mandate. Moreover, the EAC Compendium, developed in 2014 by the EAC-MRH TWG to provide guidance to the NRAs in managing applications, needs to be revised, as it is now 10 years since these guidelines were developed [22].
3.4.3 Set Number of Cycles for the Review ProcessApplicants may be slow in responding to queries, thereby delaying the review process, and there is a need to review the query response cycles (round of queries). Currently, four cycles are permitted; however, it is important to reduce the maximum to two query cycles, after which the application should be removed from the process and resubmitted as a new application (Fig. 9). A guideline on time points should be developed and implemented. The NRA time points should be evaluated when all requirements for registration are available, and it is important for metrics to include only regulators’ time at this point so that it is clear on how long regulators take to review a product. The time points for manufacturers to respond to queries should be 60 calendar days for the first queries and 30 calendar days for the second as per the example from the SADC-MRH pilot on a centralised procedure. Furthermore, the manufacturers’ response to the queries from the region review should be sent to all member NRAs. Subsequently, the individual NRAs should market the products that have been jointly reviewed on receiving the regional recommendation. However, if the manufacturer only wishes to market in one or two member estates, they should submit individually to the respective NRAs and not use the regional process.
Fig. 9
Current evaluation process. East African Community report cycle
3.4.4 Conduct Stakeholder Consultations for the EAC Work SharingIt is important to conduct stakeholder consultations in order to attract more applications. It would be helpful to perform online webinars to attract new applicants and to create an awareness of the joint review sessions as well as prepare and share expressions of interest for applicants to submit applications for the joint review. In addition, a coordinating point for country-level engagements should be established and validating exercises conducted to ensure clean and accurate data for each application that is approved available in that country.
3.4.5 Capacity Building and Training of AssessorsOne recommendation is to use the WHO Competency Framework to evaluate the ability of the assessors and identify training needs. It is difficult to track the impact of the training offered to assessors over the years as this has not been monitored and assessors attend training on an ad hoc basis. Each REC MRH should develop a list of training needs for the year. which will be handed to the Regulatory Capacity Development Technical Committee of the AMRH/AMA, who will then coordinate this training, using existing Regional Centres of Regulatory Excellence (RCORE), as well as other training opportunities that are available. It should be noted that a certification scheme for assessors has been established by the University of Witwatersrand, and it is currently sponsored by the Gates Foundation [23].
3.4.6 Develop Website and Implement a Regulatory Information Sharing PortalThe MRH programme should publish all recommended products on their websites and implement the AMRH regulatory information sharing portal (RISP) project that will enable them to share regulatory information and knowledge exchange on the continent. An electronic document management system (EDMS) is being developed through RISP that will also assist the RECs MRH to manage applications received and the distribution of the application to the assessors for preliminary review before the joint review meetings are organised.
As indicated in Fig. 10, the RECs information management system will be the interphase between national and continental RIMS. It is important that the EAC-MRH develop a robust information management system that will implement the continental digitalisation strategy at the REC level. The activation and updating of the EAC website to advocate joint activities should also be implemented.
Fig. 10
The guiding principles of the continental (African Medicines Regulatory Harmonisation/African Medicines Agency) review process. EMP-TC Evaluation of Medical Products Technical Committee, REC regional economic community
The additional weaknesses and challenges found in the current operating model of the initiative, such as the lack of detailed information for applicants on procedures and the inadequate tracking and monitoring of timelines for products in the participating countries once the joint review is completed, should be addressed by an investment in robust information management systems. By giving the region the authority to disclose this information for interested parties, this investment will increase process openness and confidence. Additional research should be carried out in these areas, which will promote increased transparency and the use of metrics to increase efficiency. It is important to have a centralised online system to make it easier for the applicants to track their applications and indicate whether they wish to follow a joint or country process. In addition, the AUDA-NEPAD, Trademark Africa, and TMDA IT experts should align efforts to link the metrics used for the EAC-MRH process to the RISP, which is currently under development.
The EAC-MRH should improve the metrics currently being collected. Also, the EAC Secretariat should recruit a biostatistician who can continue to improve the processes for capturing timelines and ensuring understanding.
3.4.7 Communication with ApplicantsAs an initiative that implements a decentralised procedure, EAC-MRH should inform both current and potential applicants at time of submission of the target timelines for the joint review process and emphasise that, similar to other decentralised procedures like those of the European Medicines Agency (EMA) or Australia-Canada-Singapore-Switzerland Consortium (ACCESS), approval timelines in different countries will vary and depend on the national process. Also, the EAC-MRH should implement the practice of publishing an expression of interest as is the situation with the ECOWAS MRH.
3.4.8 Define Roles and Responsibilities of the EAC-MRH in the AMA EraAccording to the AMA Treaty, RECs have a fundamental role to play in the regulatory ecosystem in Africa. There are three levels of this ecosystem (national—NRA; regional—REC; continental—AMA), each of which will need defined roles and responsibilities to avoid duplication. The roles of the RECs in the three-tier medicine regulatory system as recommended would include promoting collaboration; coordinating on-going AMRH activities within the region; including regulatory responsibilities for selected activities and supporting NRAs lacking capacity in identified activities; vigilance of products, especially against the movement of substandard and falsified products; providing guidance; providing links between AMA and NRAs; organising joint evaluations, inspections, and other such activities; designation, promotion, strengthening, coordination, and monitoring of RCOREs; and coordinating the collection, management, storage, and sharing of information on medical products, including substandard and falsified medical products.
From the roles and responsibilities highlighted above, a key recommendation from this study is to define a minimum functional package of structure, infrastructure, human resources, policies, and communication that would enable the EAC-MRH to be the gateway for AMA implementation. The suggested priority categories by the AMRH TC on Evaluation of Medicinal Products (EMP TC) for medical products for the continental review is illustrated in Fig. 11.
Fig. 11
Priority categories for continental review of medicinal products in Africa (EMP TC)
3.5 Part 3: A Proposed New Improved Model for the EAC-MRH InitiativeBased on the outcomes of this research, the key challenge identified that has negatively affected the effectiveness and efficiency of this initiative is the lack of a centralised process for the submission and tracking of dossiers. It is therefore recommended that a centralised submission process be implemented for the EAC-MRH as a new improved model for this initiative (Fig. 8). This will eliminate most of the challenges identified in this research and give the EAC-MRH Secretariat a legal mandate to receive and review applications. This will entail the establishment of a Regional Medicines Authority for the EAC. The review process should be simplified and predictable, with proposed timelines that will make the process more attractive over the standard pathway. The guidance on using this centralised process should be a “SMART” initiative especially with the introduction of an electronic process (e-CTD). A centralised process for the payment of fees for joint reviews should be established alongside this process. Instead of having too many entry points, applicants interested to have their applications reviewed through the EAC-MRH should apply directly to the EAC-MRH, after which the review process as per Fig. 8 can start. Health is for all, and this study proposes that manufacturers who choose to use the regional joint review process should submit their applications to all NRAs in the community. This will increase access to medical products, which is the goal of the initiative rather than manufacturers selecting only certain member states and not others. If a manufacturer only wants to market in one of two member states, they should submit individually to those NRAs and not use the regional process.
Milestone one will then be the recording of the date at which the application and screening fees are received (step 1). The centralised submission will eliminate the 7-day deadline given to the countries to submit the applications they have received by the Lead Authority. Instead, screening of the application should be carried out within 5 days after receipt of the application. Screening fees should also be paid during the time of submission of the application. In step 2, the EAC-MRH Secretariat would screen and validate the application. If there is missing information, the applicant would be notified and additional information submitted within 5 days. The EAC-MRH would then assign the application for an initial review by the first assessor
Comments (0)