
Remember me
Dear Editor,
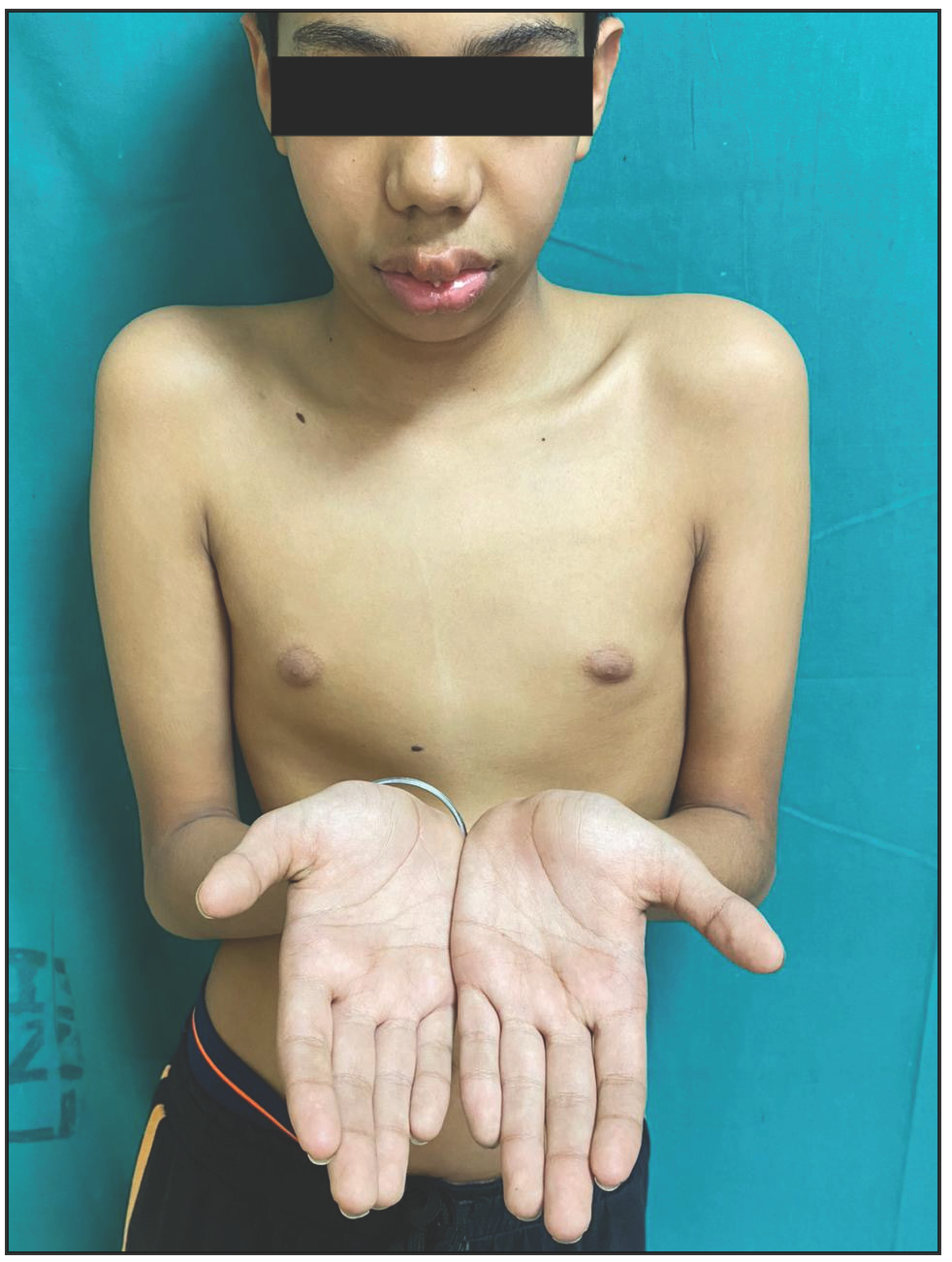
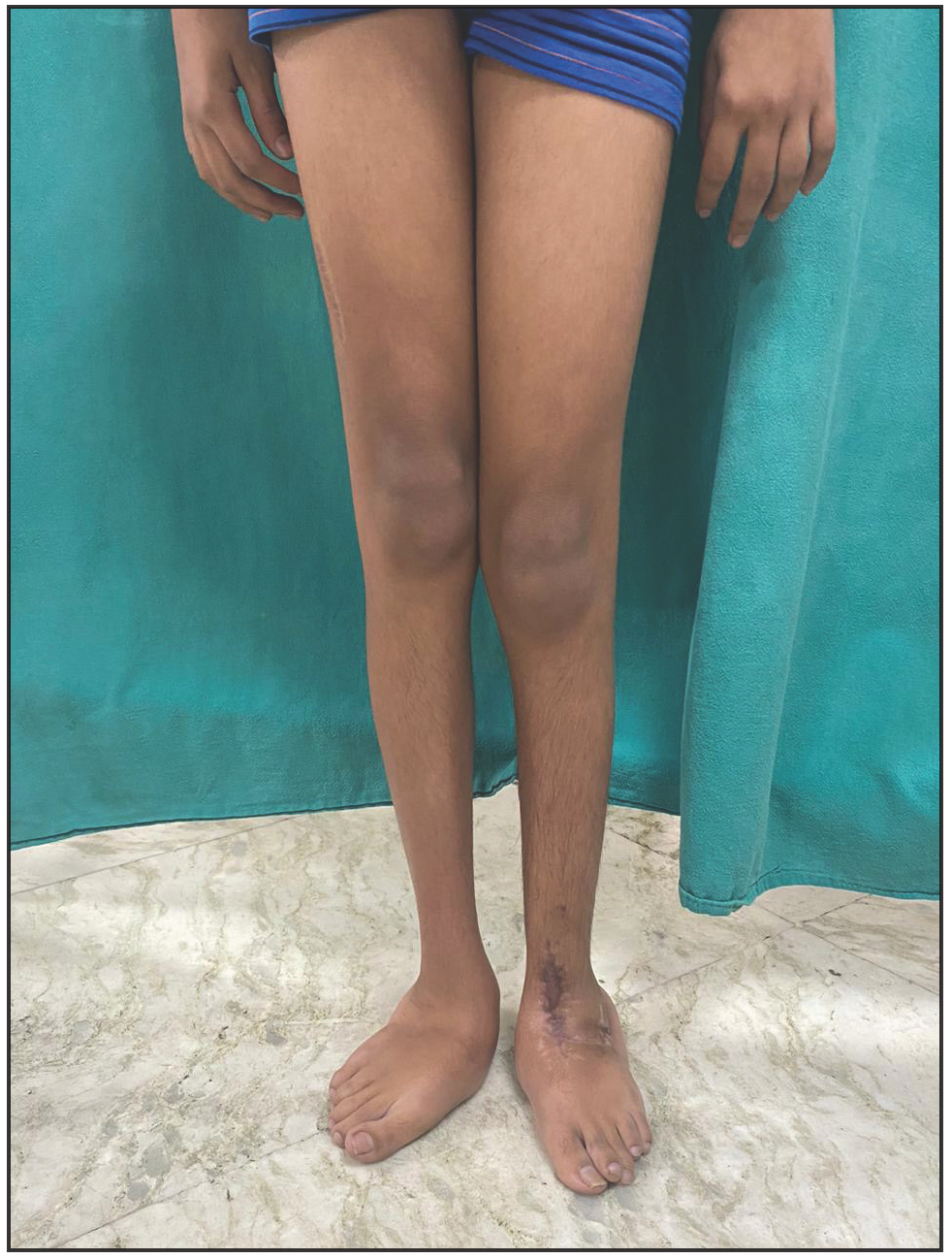
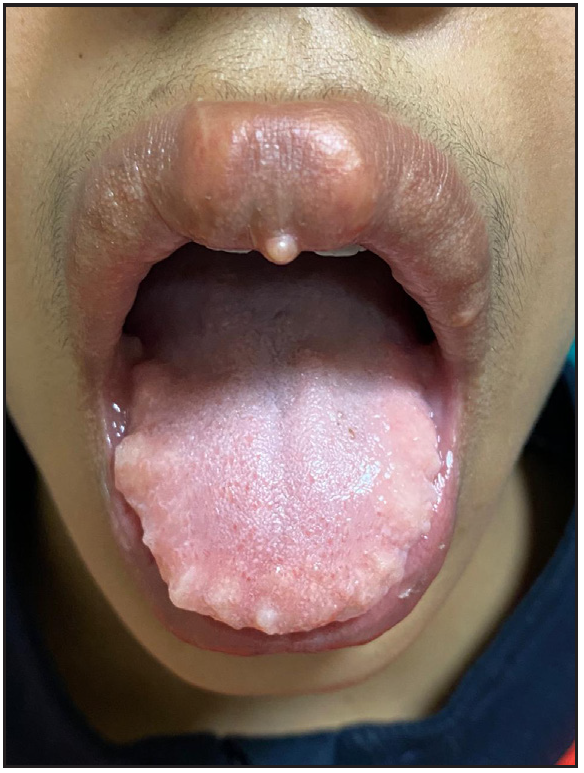
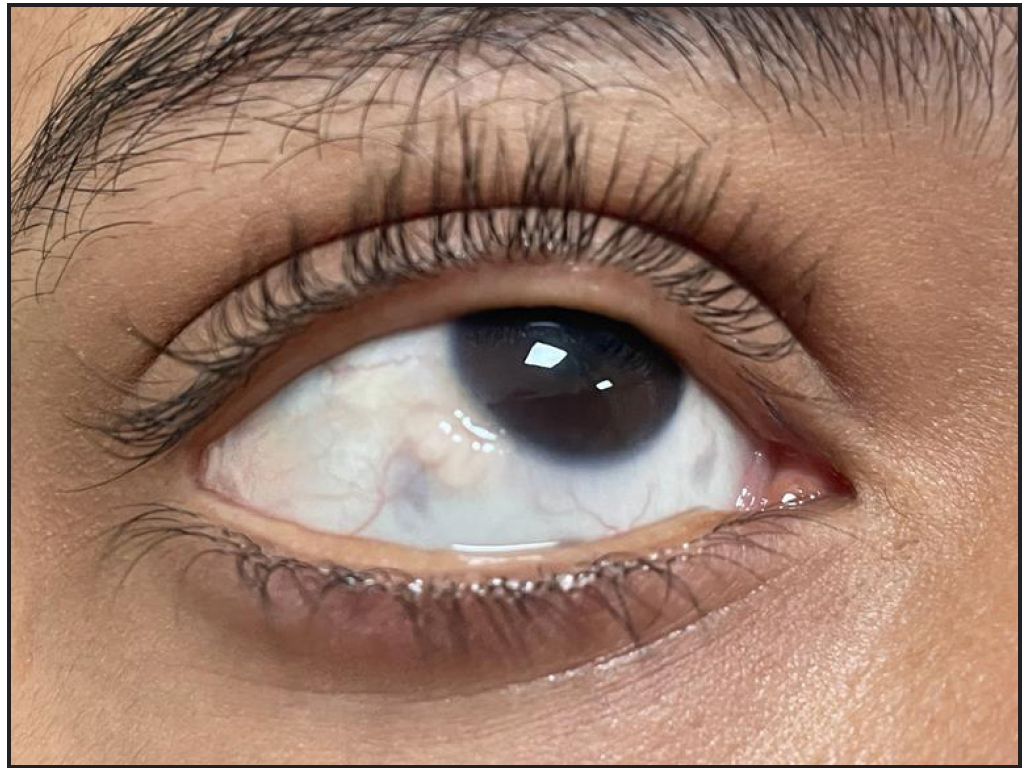
A 12-year-old boy presented with swelling of the lips and yellowish nodular lesions on the tongue and lips. These lesions were persistent, asymptomatic, and gradually increasing in size and number since the age of one-year. On examination, he appeared thin and slender and had disproportionately longer arms and legs than the trunk, pointing towards the marfanoid habitus [Figures 1a and 1b]. The lips were large, everted, and had an uneven appearance due to the presence of multiple yellowish nodules [Figure 2a]. Similar lesions were also noticed on the margins of the tongue and anterior buccal mucosa. A single, well-defined, gelatinous, irregularly shaped, conjunctival nodule measuring 3 mm in size was noticed over the scleral portion of the right eye, suggestive of a conjunctival neuroma [Figure 2b]. Musculoskeletal examination revealed long fingers, hallux valgus, scoliosis, and a tilted pelvis. His previous medical records showed talonavicular reposition surgery for congenital vertical talus with planovalgus deformity of his left foot. He also had thick eyebrows and eyelashes. A biopsy of one of the mucosal nodules was sent for histopathological examination, which revealed multiple spindle-shaped Schwann cells in the submucosa, arranged in a tortuous, unorganised manner, suggestive of mucosal neuroma [Figure 3].
Export to PPT
Export to PPT
Export to PPT
Export to PPT
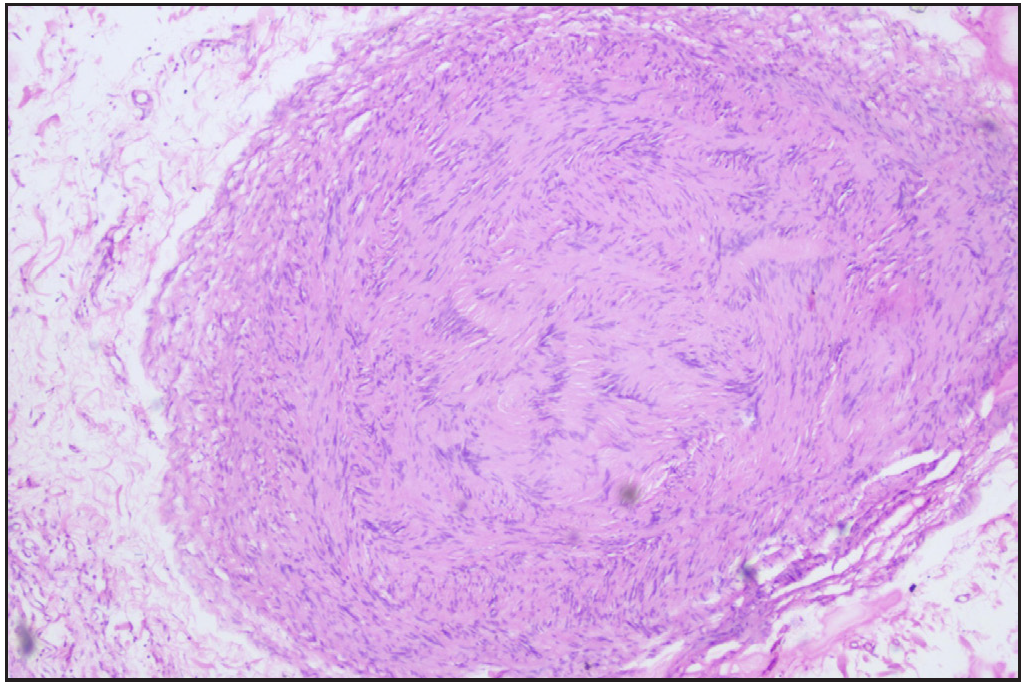
Export to PPT
The constellation of features of mucosal and conjunctival neuromas, marfanoid habitus, and bony abnormalities lead to a diagnosis of multiple endocrine neoplasia type 2B (MEN2B) syndrome. However, genetic testing for RET protooncogene mutation could not be performed due to a lack of resources. No family history of similar complaints or malignancies was present. The thyroid gland was enlarged without any definitive nodules. There was no peripheral lymphadenopathy. Serum and 24-hour urinary metanephrines were negative, and blood pressure was normal, ruling out pheochromocytoma. An ultrasonographic examination of the neck was advised by the surgeon and it revealed the presence of benign modules (Thyroid Imaging Reporting and Data System/TIRADS 3). However, due to the high chances of medullary carcinoma thyroid in MEN2B syndrome, a prophylactic thyroidectomy was done.1 Currently, he is under regular follow-up and screening for pheochromocytoma in the surgery department.
Many times, hereditary cancer syndromes have associated cutaneous features, which develop early in life and hence aid in the early detection of underlying sinister malignancies. Knowledge regarding these signs enables the dermatologists to establish a diagnosis and refer the patient to the oncologists, thereby preventing life-threatening complications. Mucosal neuromas, although typically seen in MEN2B syndrome, may be missed due to a lack of awareness. MEN2B, also known as Wagenmann-Froboese syndrome, is caused by activating mutations in the RET-proto-oncogene. These individuals are prone to develop medullary thyroid carcinoma and pheochromocytoma, resulting in mortality. Hence, screening at an early age is recommended.1 Total thyroidectomy at an early age can cure or prevent medullary thyroid carcinoma and prolong life.2 Patients with MEN2B syndrome have typical cutaneous and musculoskeletal features that help in its diagnosis, especially in cases without family history. The typical cutaneous feature of this syndrome is the presence of mucosal neuromas on the lips, tongue, and buccal mucosa and everted prominent thick lips, which are seen in almost all cases. Mucosal neuromas are characterised by yellowish asymptomatic soft to firm pedunculated or sessile masses, manifesting at birth or during early childhood. Similarly, conjunctival neuromas involving the conjunctiva and enlarged medullated corneal nerve fibres are commonly seen.3 Other less frequently observed cutaneous features include cutaneous neuromas (perinasal), perioral pigmentation, and hypertrichosis. Marfanoid habitus, kyphoscoliosis, pes planus/cavus, pectus excavatum, congenital hip dislocation, proximal myopathy, and reduced subcutaneous fat are the associated musculoskeletal features. Synorphys, high arched palate, broad forehead, protruded mandible, elongated face, and hypertelorism may be noticed in a few cases.4 A summary of this disease with its clinical features and screening protocol is given in Table 1. Frequently, dermatologists may be the first to encounter patients with MEN2B syndrome due to prominent cutaneous signs, as seen in our case. Hence, through our case, we want to highlight the importance of a dermatologist in recognising the cutaneous signs, especially neuromas, which can help in the early diagnosis and multidisciplinary management of this syndrome, involving a team of surgeons, endocrinologists, oncologists, and genetic counsellors.
Table 1: Summary of MEN2B syndrome
Aspect Details AetiologyCaused by activating mutations in the REarranged during Transfection (RET)-proto-oncogene located on chromosome 10.
Autosomal dominant inheritance.
Cutaneous featuresMucosal neuromas: Yellowish, asymptomatic, soft to firm sessile, or pedunculated nodules (lips, tongue, buccal mucosa, pharynx, larynx, conjunctiva).
Perinasal cutaneous neuromas
Hypertrichosis, perioral pigmentationEverted, thick lips, eyelids.
Extra-cutaneous featuresMusculoskeletal: marfanoid habitus, kyphoscoliosis, pectus excavatum, long fingers, hip dislocation, pes planus/cavus
Proximal myopathy, hypotonia, and reduced subcutaneous fat may be seen.
Typical facies: synorphys, high arched palate, broad forehead, protruded mandible, elongated face, and hypertelorism Ocular: conjunctival neuromas, thick corneal nerves, and thickened eyelidsEndocrine: medullary thyroid carcinoma, pheochromocytoma, hyperparathyroidism.
Ganglioneuromatosis of the digestive tract
Autonomic manifestations- impaired lacrimation, orthostatic hypotension, impaired reflex vasodilation of the skin, parasympathetic denervation super-sensitivity of the pupils
Dental manifestations- increased interdental space
Screening and follow-up protocols Genetic testing for RET mutations and appropriate counselling. Annual biochemical screening for phaeochromocytoma is necessary for all with RET mutations or in a MEN2 kindred with no identified mutations. A 24-hour urinary excretion of normetanephrine & metanephrine is the most sensitive test. A positive test should prompt imaging (CT/MRI and, if positive, MIBG scanning). A positive scan is an indication for surgery.Calcitonin is the tumour marker for medullary thyroid carcinoma and should be assayed before thyroidectomy if possible, and then annually thereafter. During follow-up, a rising calcitonin or a palpable mass are indications for imaging. Ultrasound and CT or MRI are suitable structural imaging modalities, while sestamibi scanning may localise functioning tissue. MIBG scanning may occasionally be useful where other radiopharmaceuticals fail to image. Prophylactic thyroidectomy in early childhood can be done to prevent medullary thyroid carcinoma. Differential diagnosisMarfan syndrome (no neuromas, no endocrine tumours). MEN2A (endocrine tumours present; no marfanoid habitus; cutaneous features include lichen amyloidosis and notalgia paresthetica).
Cowden syndrome (neuromas present but marfanoid habitus absent, typical cutaneous features are trichilemmomas, acral keratoses, and oral papillomas) Neurofibromatosis type 1(cutaneous neurofibromas, café-au-lait spots).
Prognosis Early diagnosis and intervention (e.g., thyroidectomy) significantly improve survival by preventing metastatic medullary thyroid carcinoma, otherwise high risk of early mortality at a young age.
Comments (0)