Patients and specimens
The diagnostic criteria for patients with RA were based on the American College of Rheumatology criteria [26]. The diagnostic criteria for patients with OA were based on the classification criteria from the ARC in 1986. Synovial tissues from patients with OA or RA who underwent total knee replacement were obtained. Normal synovial tissues were obtained from young patients with meniscus injury or cruciate ligament injury who underwent arthroscopic surgery. Ethical approval was obtained from the Ethics Committee of Henan Provincial People’s Hospital (Zhengzhou, China, LLXJS2023-1-137). A total of 12 RA patients (7 females, 5 males; mean age 56.8 ± 4.1 years), 11 OA patients (6 females, 5 males; mean age 55.0 ± 6.1 years), and 7 healthy controls (3 females, 4 males; mean age 26.7 ± 2.2 years) were enrolled in the study. The disease duration was 8.5 ± 3.2 years for RA patients and 10.2 ± 4.1 years for OA patients. Written informed consent was obtained from all participants.
Cell isolation and culture
The synovial tissues from patients were washed with PBS under sterile conditions. Then, fat tissues were carefully removed with ophthalmic scissors, and the tissues were cut into small pieces. The tissues were digested with 2.5% type I collagenase (Biosharp, Hefei, China) for 2 h in a constant-temperature shaker. Single-cell suspensions were subsequently prepared through a cell strainer. The cells were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (Hangzhou Sijiqing, China) and incubated at 37 °C with 5% CO2. The cells were used after passages 3–5 for subsequent experiments.
Identification of differentially expressed circrnas
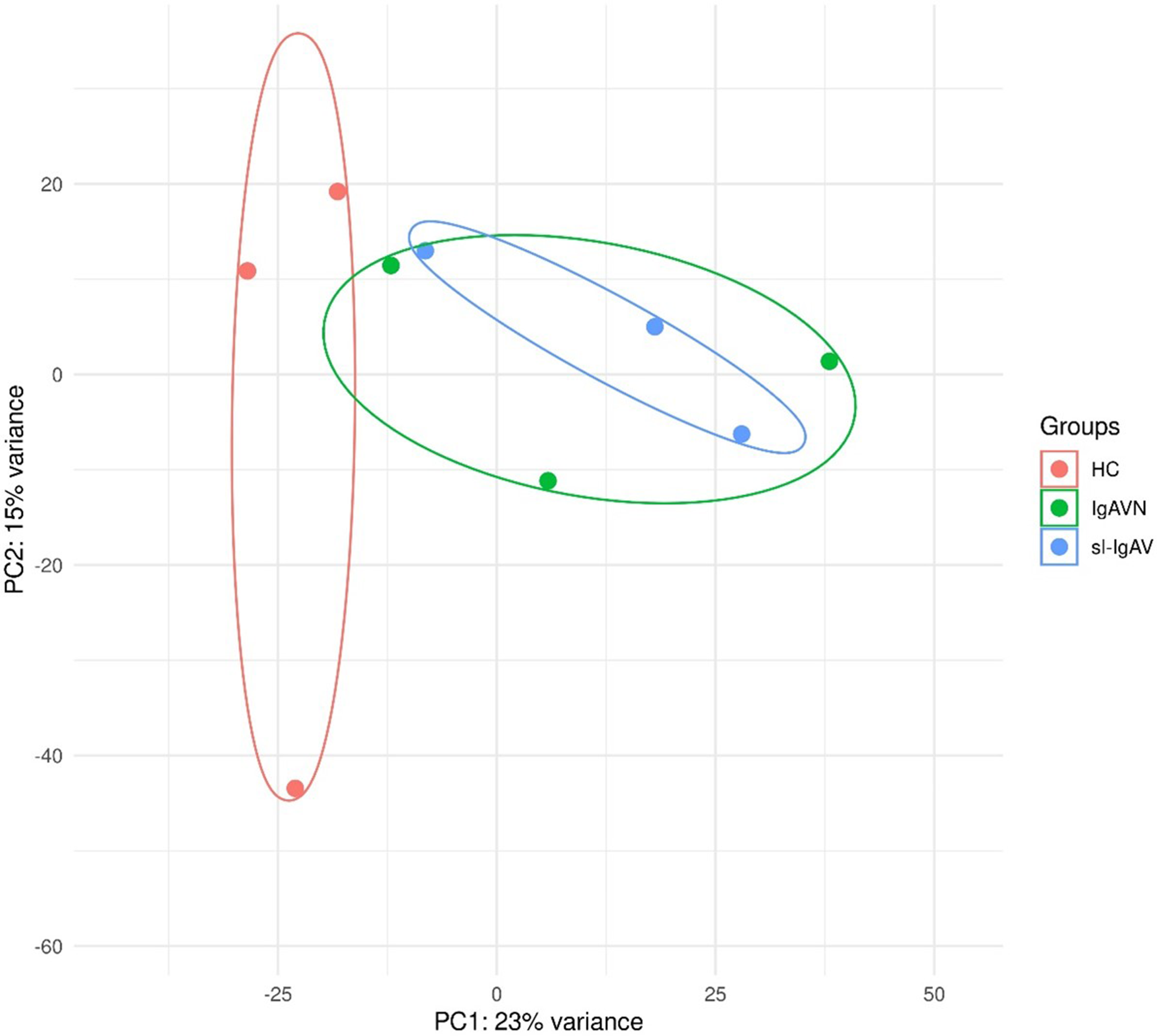
FLSs were isolated from three healthy controls, three OA patients, and three RA patients. The total RNA of FLSs was collected for circRNA sequencing. The circRNA library was constructed via rRNA removal and linear RNA digestion. Purified and enriched circRNAs were fragmented and used for cDNA synthesis to construct the library. The purified double-stranded cDNA was subsequently subjected to end-repair, dA-tailing and adaptor ligation. The cDNA products were circularized and amplified via PCR and subsequently used for DNB generation. Finally, each DNB was loaded into a lane for subsequent sequencing on a BGISEQ-500 platform (MGI, Shenzhen, China). The circRNA candidates were detected by using find_circ, as abnormal fragments were discarded [21]. CircRNAs with a|log (fold change)| >2 and adjusted P value < 0.05 were set as the significant cutoffs. Gene Ontology (GO) analysis and enrichment analysis of these differentially expressed circRNAs were performed via Sanger box 3.0.
RNase R treatment
To assess the stability of circRNAs, linear mRNA was digested with RNase R (Beyotime). The RNase R treatment was performed in a 20 µL reaction system, which included ≤ 5 µg of total RNA, 2 µL of 10X RNase R Reaction Buffer, and RNase R (20 U/µL) at a final concentration of 1–3 U per µg of RNA. The reaction volume was adjusted to 20 µL using DEPC-treated water. The mixture was then incubated at 37 °C for 30 min to allow complete digestion of linear RNA. After treatment, the stability of the circRNAs was determined by qRT-PCR.
Sanger sequencing
Further verification of the accuracy of circRNA primer amplification was performed via Sanger sequencing by Company. (Shenzhen, China)
Nucleic acid transfections
RA-FLSs were grown in 12-well plates and reached 60-80% confluence for subsequent transfection. siRNAs against hsa_circ_0002970 and negative control siRNA were purchased from Geneseed Technology (China). The target sequences for siRNA-mediated knockdown of circ_0002970 were as follows: (1) Si-hsa_circ_0002970-1: CAATGGCTTGTTGGGAAAC(dT)(dT); (2) Si-hsa_circ_0002970-2: AATGGCTTGTTGGGAAACC(dT)(dT); (3) Si-hsa_circ_0002970-3: ATGGCTTGTTGGGAAACCA(dT)(dT); (4) Si-NC: UUCUCCGAACGUGUCACGUTT(dT)(dT).The negative control siRNA that was labeled with FAM was used to visualize the uptake of the siRNA. The transfection was performed by using Lipofectamine® RNAiMAX Reagent (Thermo Fisher Scientific). Opti-MEM reduced serum medium was used to dilute the siRNAs, and Lipofectamine® RNAiMAX Reagent was used.
Lentiviral transfection
Lentiviral vectors for circ_0002970 overexpression (LV-circ_0002970) and negative control (LV-NC) were designed and synthesized by HANBIO (Shanghai, China). RA-FLSs were seeded in six-well plates and cultured until they reached 50–70% confluence. The cells were then infected with lentivirus at an appropriate multiplicity of infection (MOI) in the presence of 5 µg/mL polybrene to enhance transduction efficiency. After 24 h, the medium was replaced with fresh complete DMEM, and cells were further cultured for 48–72 h. For stable expression, cells were selected using 2 µg/mL puromycin for 5–7 days.
ELISA kit
An IL-6 human ELISA kit (Ruixin Biotech, QuanZhou, China) was used. RA-FLSs were plated on 6-well plates. After circ_0002970 was knocked down, the cells were incubated with DMEM containing 10% fetal bovine serum for 24 h. The supernatant was collected to measure the IL-6 levels.
Migration and invasion assays
The migration and invasion abilities of FLSs were assessed using 24-well Transwell chambers with 8 μm pore-size polycarbonate membranes (Corning). For the invasion assay, Matrigel was pre-diluted on ice at a ratio of 8:1 (FBS: Matrigel) and evenly coated onto the upper surface of the Transwell insert. The coated chambers were incubated at 37 °C for 4–5 h to allow polymerization. For the migration assay, no Matrigel coating was applied.After Matrigel polymerization, 200 µL of a serum-free DMEM cell suspension containing FLSs was added to the upper chamber, while 600 µL of DMEM supplemented with 5% FBS was added to the lower chamber to create a chemoattractant gradient. The cells were incubated at 37 °C in a humidified incubator with 5% CO2 for 24 h to allow migration (or invasion).Following incubation, the non-migrated or non-invaded cells on the upper surface of the membrane were gently removed using a cotton swab. The membranes were then fixed with 4% paraformaldehyde for 15 min and stained with crystal violet solution for 15 min. The stained cells were visualized using an inverted microscope, and the number of migrated or invaded cells was counted in four randomly selected fields (×200 magnification).
RNA labeling and fluorescence in situ hybridization
FISH analysis of RA-FLSs was performed using biotin-labeled probes specific to circ_0002970 (Servicebio, Wuhan, China). RA-FLSs were cultured on glass coverslips and then fixed with 4% paraformaldehyde. The cells were permeabilized with 0.1% Triton X-100, followed by prehybridization at 37 °C. Biotin-labeled probes specific to circ_0002970 were hybridized overnight at 37 °C. After posthybridization washes with 2x and 0.5x SSC buffer, the probes were detected via a fluorescently labeled streptavidin conjugate. Nuclei were counterstained with DAPI, and the coverslips were mounted on slides with antifade medium. Finally, cell observation and fluorescence imaging were performed via a fluorescence microscope (Nikon, Tokyo, Japan).
Agarose gel electrophoresis
The qPCR products amplified by convergent primers and divergent primers were subjected to 1.5% agarose gel electrophoresis. First, the agarose was diluted with Tris-acetate-EDTA buffer in a erlenmeyer flask and then heated in a micro-wave oven. Then, the cDNA and gDNA qPCR products were mixed with loading buffer added to the agarose gel well, a suitable ladder was chosen and separated at 110 V for 30 min. The gel was soaked in ethidium bromide to stain the DNA fragments and visualized in an automatic gel imaging system (DYY-8c, Beijing Liuyi Co., Ltd., Beijing, China).
Quantitative real-time PCR validation
The total RNA of FLSs was extracted via the FastPure Cell/Tissue Total RNA Isolation Kit (RC112, Vazyme Biotech). The gDNA of FLS was extracted via the FastPure® Cell/Tissue DNA Isolation Mini Kit (DC102, Vazyme Biotech). Then, qRT‒PCR was performed via a ChamQ Universal SYBR qPCR Master Mix Kit (Q711, Vazyme Biotech) in a 7500 Fast Real-time quantitative PCR system according to the standard procedure (Thermo Fisher Scientific). The relative gene expression of the circRNAs and mRNAs was analyzed via normalization to the expression of GAPDH via the 2-ΔΔCt method. The linear and circular RNA primers used in this study were synthesized by Shangya Biotechnology Co., Ltd. (Supplementary material 1).
HE staining
The synovial tissues were excised from the knee joints, fixed in 10% neutral formalin, decalcified in EDTA, embedded in paraffin, sectioned, and stained with hematoxylin‒eosin (HE).
Western blotting
FLSs were grown in 6-well plates and treated differently (2 × 105/well). Then, the cells were lysed in a mixture of RIPA lysis buffer (Thermo Fisher Scientific) with 1 tablet of protease inhibitor and 1 tablet of phosphatase inhibitor (MedChemExpress) for protein extraction. A BCA protein assay kit (Beyotime, China) was used for protein concentration determination and standardized quantification. SDS‒PAGE was used to resolve the proteins, which were then transferred to PVDF membranes. Western blotting was carried out according to standard procedures. The membranes were incubated with specific antibodies and then visualized via ChemiDoc XRS (Bio-Rad). To ensure reproducibility, WB experiments were performed with biological triplicates (n = 3 per group, each representing an independent sample).
Statistical analysis
All the experiments were repeated at least three times. GraphPad Prism 9 and SPSS 26.0 software were used for statistical analysis. All the data are presented as the means ± standard deviations. Differences between two groups were analyzed via Student’s t test, whereas differences among three or more groups were analyzed via one-way ANOVA. P < 0.05 was considered statistically significant.








Comments (0)