
Remember me

Prenylemestrin C (1) was isolated as a white powder with the molecular formula C32H32N2O11S2, based on the HRESIMS spectrum with an ion peak at m/z 707.1345 ([M + Na]+, calcd. for 707.1345), indicating 18 degrees of unsaturation. The IR spectrum revealed the presence of hydroxyl (3419 cm−1), carbonyl (1691 and 1662 cm−1), and aromatic ring (1513 cm−1) functionalities. The 1H NMR data of 1 recorded in CD3OD (Table 1) showed signals corresponding to two 1,3,4-trisubstituted benzene rings protons at δH 7.98 (d, J = 2.0 Hz), 7.81 (dd, J = 8.6, 2.0 Hz), and 7.19 (d, J = 8.6 Hz), and δH 8.42 (d, J = 2.3 Hz), 7.28 (dd, J = 8.6, 2.3 Hz), and 6.90 (d, J = 8.6 Hz), seven methines including three olefinic [δH 6.92 (d, J = 2.4 Hz), 6.42 (dd, J = 8.1, 2.4 Hz), and 5.01 (dd, J = 8.1, 2.3 Hz)] and three oxymethines [δH 5.49 (dt, J = 8.3, 2.3 Hz), 5.12 (s), and 4.72 (s)], one methylene [δH 3.46 (dd, J = 16.5, 1.8 Hz) and 2.89 (dd, J = 16.5, 5.3 Hz)], a methoxy group (δH 3.99), an N-methyl (δH 3.36), and two singlet methyls (δH 0.80, 0.68). The 13C NMR and DEPT spectroscopic data of 1 recorded in CD3OD displayed 32 carbon signals (Table 2), including three carbonyls (δC 167.2, 167.1, 161.5), 10 nonprotonated carbons (seven olefinic and three oxygenated sp3 at δC 78.0, 76.5, 73.8), 14 methines (nine olefinic and five sp3), a methylene (δC 35.3), and four methyls (δC 56.8, 29.0, 26.1, 28.4).
Table 1 1H NMR Spectroscopic Data (400 MHz; δ in ppm, J in Hz) for Compounds 1–5Table 2 13C NMR Spectroscopic Data (100 MHz; δ in ppm) for Compounds 1–5The planar structure of compound 1 was elucidated through 2D NMR spectral analysis. The independent spin system of H-5a/H-6/H-7/H-8, along with HMBC correlations of H-10/C-5a, C-8, and C-10a, confirmed the presence of a 6,7-dihydrooxepine ring. Further HMBC spectrum showed correlations from H-11 to C-5a, C-10a, C-11a, and C-1, as well as from H-5a to C-4 and from the N-methyl protons to C-1 and C-3. These findings established that the 6,7-dihydrooxepine ring was fused to a dioxopiperazine moiety via an 11-hydroxypyrrolidine ring. Additionally, a 3′-oxygen-4′-methoxy-benzoate fragment and a 3′′-oxygen-4′′-hydroxybenzyl unit were identified as being connected to the 6,7-dihydrooxepine ring at C-6 and the dioxopiperazine ring at C-3, respectively. This connectivity was supported by the HMBC correlations from H-6 to C-7′ and from H-7′′ to C-3, C-4, C-1′′, C-2′′, and C-6′′. Moreover, the 1H−1H COSY correlation of H2-12/H-13, combined with the HMBC cross-peak from H2-12 to C-11a, H-13 to C-3, H3-15/H3-16 to C-13 and C-14, confirmed the attachment of an isopentan-3-ol fragment to C-11a and C-3 through sulfur atoms. Consequently, compound 1 was deduced to feature an epipolythiodioxopiperazine skeleton with a thioethanothio bridge, closely resembling the planar structure with prenylemestrins A and B (6 and 7) [17]. A detailed comparison of their 1H and 13C NMR data revealed that the key difference lies in the 2,5-dithia-7,9-diazabicyclo[4.2.2]decane-8,10-dione core. HMBC correlations (Fig. 2) from H2-12 to C-11a and from H-13 to C-3 confirmed that C-12 and C-13 were linked to C-11a and C-3, respectively. With the hemiterpene moiety switched, 1 can also be interpreted as a migration of the 2-hydroxyisopropyl group.
Fig. 2
Key 1H−1H COSY and HMBC correlations of compounds 1−5
To determine the relative configuration of compound 1, NMR data were recorded in DMSO-d6 (Tables 1 and 2). The trans-orientation of H-5a and H-6 was confirmed by the large coupling constant (J = 8.3 Hz) observed between those protons [21]. The NOESY correlation between H-5a and OH-11 indicated that H-11 and H-6 were on the same side, tentatively assigned as α orientation [17]. Furthermore, NOESY correlations (Fig. 3) of H-6/H-13, H-2′/Me-15/Me-16, H-2″/Me-15/Me-16, suggested that the configuration of C-13 should be R*. Accordingly, the C-3 − S and C-11a − S bonds were deduced to be α-oriented. Additionally, NOESY interactions of N-Me/H-7″, H-7″/H-2″, and H-7″/H-6″, OH-7″/H-6″, and OH-7″/N-Me, supported the assignment of the configurations at C-7′′ as S*. Based on these observations, the relative configuration of 1 was established.
Fig. 3
Key NOESY correlations of compounds 1−5
Prenylemestrin D (2) was determined to share the same molecular formula, C32H32N2O11S2, as compound 1, based on HRESIMS and NMR spectra. A comparison of the 1D (Tables 1 and 2) and 2D NMR spectra (Fig. 3) revealed significant similarities between the two compounds, except for the slightly different chemical shifts of C-1 (2: δC 170.4; 1: δC 167.1), C-4 (2: δC 165.4; 1: δC 161.5), and C-11a (2: δC 75.3; 1: δC 78.0), which inferred that 2 could be the C-13 epimer of 1 (Fig. 1). This deduction was further supported by NOESY interactions of H-13/N-Me and H-6/H-12a (δH 2.76) in 2. Additionally, the experimental ECD spectra (Fig. 4) of compounds 1 and 2 closely matched those of 6 and 7, whose absolute configurations had been previously confirmed by X-ray diffraction (Fig. 5). These findings conclusively established the absolute configurations of 1 and 2 as depicted in Fig. 1.
Fig. 4
Experimental ECD spectra of compounds 1, 2, 6, and 7
Fig. 5
ORTEP drawing of the X-ray crystal structures of 6 and 7
Prenylemestrin E (3) was assigned the molecular formula of C32H32N2O11S2, as indicated by its HRESIMS spectrum, showing an ion peak at m/z 707.1372 ([M + Na]+, calcd. for C32H32N2O11S2Na+, 707.1345), corresponding 18 degrees of unsaturation. The 1D NMR data of 3 closely resembled those of 1 and 2 (Tables 1 and 2), except for the appearance of distinctive signals for a conjugated aldehyde group (δC 190.9 and δH 9.75), and a methine (C-3, δH 4.96, s; δC 65.7), which replaced the nonprotonated carbon C-3 (δC 76.5) in 1. These differences suggested that 3 was structurally related to secoemestrin C, with cleavage of the 15-membered macrolactone ring between C-3 and C-7′′. HMBC correlations from H-7′′ to C-1′′, C-2′′, and C-6′′ confirmed the presence and position of an aldehyde group. Moreover, HMBC interactions from H-13 to C-3, and from H2-12 to C-11a along with the 1H − 1H COSY correlations of H2-12/H-13 further established the caged core structure of 3 as consistent with that of 1 and 2. The relative configuration of 3 was deduced from the NOESY spectrum and coupling constant analysis. The coupling constants (JH‑5a/H‑6 = 8.0 Hz) indicated that H-5a was trans-oriented. The NOESY correlation of between H-11 and H-6 indicated that H-11 is α-oriented. The observed NOESY correlation (Fig. 3) of H-6/H-13 suggested that the configuration of C-13 was R*, with the C-3−S and C-11a−S bonds being α-oriented, and H-3 being β-oriented. Furthermore, the theoretical 13C NMR calculations with DP4 + analysis of two isomers13R*-3 and 13S*-3 were conducted through the GIAO method at the mPW1PW91/6-311G(d,p) level in chloroform with the Gaussian 09 software. The results (Fig. S1) showed that 13R*-3 had a better coefficient of determination (R2 = 0.9983) between the experimental and calculated 13C NMR chemical shits than 13S*-3 (R2 = 0.9964). Additionally, the DP4 + analysis with a high probability of 100% permitted the relative configuration 13R*-3. To further confirm the absolute configuration of 3, the ECD spectra of (3R*,5aS*,6S*,11R*,11aR*,13R*)-3 were calculated using time-dependent density functional theory (TDDFT) methods (Tables S3 and S4). The calculated spectra matched well with the experimental data (Fig. 6), leading to the assignment of the absolute configurations of 3 as 3R,5aS,6S,11R,11aR,13R.
Fig. 6
Calculated and experimental ECD spectra of 3 and 5
Prenylemestrin F (4) was determined to have the same molecular formula, C32H32N2O11S2, as compound 3, as confirmed by HRESIMS and NMR data. The 1H and 13C NMR data (Tables 1 and 2) of 4 closely resembled those of 3, indicating a similar structural framework. Further interpretation of the 1H − 1H COSY and HMBC spectra (Fig. 2) confirmed that compound 4 shares the same ETPs core as 3. Notable differences between 3 and 4 were observed in the chemical shifts of C-1 and C-4, which were significantly downfield-shifted by + 4.5 ppm and + 4.9 ppm, respectively, in 3. Detailed 1H − 1H COSY and HMBC analyses confirmed that both compounds possess the same planar structure. Therefore, 3 and 4 were concluded to be stereoisomers. The NOESY interaction of H-13/N-Me was observed for 4, as opposed to H-13/H-6 in 3, which, along with careful NOESY elucidation, suggested that 4 is the C-13 epimer of 3. Furthermore, the experimental ECD spectra (Fig. 6) of compound 4 closely matched those of 3, leading to the determination of the absolute configuration of 4 as 3R,5aS,6S,11R,11aR,13S.
Prenylemestrin G (5) was assigned the same molecular formula of C32H32N2O11S2 as 3 and 4, based on HRESIMS analysis. The 1H and 13C NMR data of 5 (Tables 1 and 2) closely resembled those of 3. HMBC correlations (Fig. 2) from H2-12 to C-3, H-13 to C-11a confirmed that the connection between the hemiterpene moiety and the ETP core in 5 was consistent with prenylemestrins A and B (6 and 7) [17]. NOESY correlations of H-6/H-13 (Fig. 3) indicated that H-13 in 5 adopts an α-orientation. The absolute configuration of 5 was determined by comparison its experimental ECD spectrum with the calculated one. The calculated ECD spectrum for (3R,5aS,6S,11R,11aR,13S)-5 was in good agreement with the experimental data (Fig. 6), leading to the assignment of the absolute configuration as 5 as 3R,5aS,6S,11R,11aR,13S.
Based on previous biosynthetic studies of ETPs, hypothetic biogenetic pathways of 1−7 were proposed to explain their origins (Scheme S1) with two molecules of L-phenylalanine as precursors. The key intermediate I was obtained via a series of peptide cyclization, ring-expansion, and esterification reactions. Then, intermediate I underwent methylation and macrocyclization to form II. Sulfurization of the intermediate II at C-3 and C-11a produced the key dithiol intermediate III, which was further decorated with dimethylallyl diphosphate via pathway a or b to form III or V, respectively. Subsequently, the followed epoxidation and nucleophilic attack of the sulfhydryl group led to the production of 1−2 and 6−7. The intermediate VI, with the C-3−C-7′′ bond cleavaged, could be derived from III; ultimately, compounds 3−5 could be derived via pathways c and d, with dimethylallyl diphosphate and modified farnesyl diphosphate, respectively.
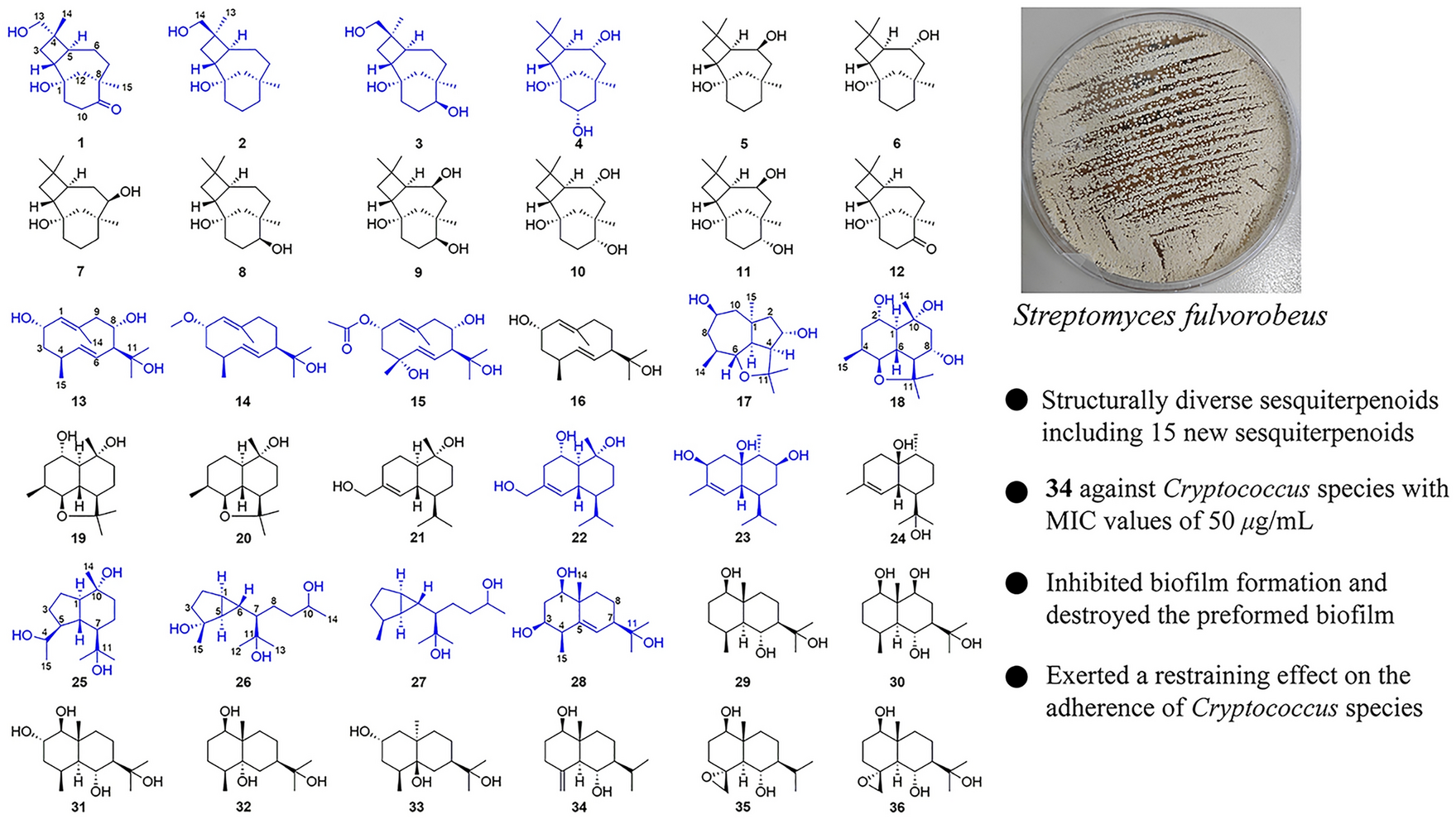
In our biological evaluation, compounds 1−7 exhibited no anti-inflammatory activity. Additionally, compounds 1−5 and 7 showed no cytotoxicity against the tested cell lines (A549, L1210, HL-60, SW-480, and Hep3B). Interestingly, 6 demonstrated moderate cytotoxicity against L1210 mouse leukemia cells. Using L1210 cells as a model, we further investigated the antitumor effect and the underlying mechanism of compound 6. The results revealed that 6 inhibited proliferation and induced G2/M cell cycle arrest in L1210 cells by regulating PI3K/AKT pathway and cell cycle-related proteins, such as p-Chk1, Cyclin B, Cdc2, p-Cdc2, and γh2ax (Fig. 7). Furthermore, 6 inhibited mitochondrial membrane potential (MMP), increased ROS levels, and induced apoptosis in L1210 cells (Fig. 8). These changes in MMP and ROS levels suggest that the antitumor effects of 6 are closely related to mitochondrial dysfunction. Western blot analysis further supported this hypothesis, showing that 6 altered the expression of mitochondria-related proteins, including Bak and Bcl-xl, indicating that apoptosis induced by 6 occurs predominantly via the mitochondrial pathway. The antitumor effects of compound 6 were also validated in other leukemia cell lines, with results consistent with those observed in L1210 cells (data not shown). These findings highlight the potential of 6 as a candidate for anti-leukemia research, warranting further in-depth studies on its mechanism and therapeutic potential.
Fig. 7
Compound 6 inhibited proliferation and induced G2/M cell cycle arrest in L1210 cells. Morphologic change of L1210 cells after treatment with 6 (A); Cell proliferation curve of CCK-8 viability assay (B); EdU assay showed that EdU positive ratio was affected by 6 treated (C). Cell cycle distribution (D); Western blot analysis of PI3K/AKT and cell cycle related proteins, with GAPDH was used as loading control (E)
Fig. 8
Effect of compound 6 on L1210 cells. Treat L1210 cells with different concentrations of 6 for 24 h and detect mitochondrial membrane potential status using JC-1 staining kit (A−C). D Fluorescence data of DCFH-DA staining of cells treated with 6 or Rosup for 36 h. E Cell apoptosis was determined by flow cytometric analysis using Annexin V-FITC and PI staining. F Western blot analysis of apoptosis related proteins, with GAPDH used as loading control
Comments (0)