Animal model and treatment
All animal experiments conducted in this study received approval from the Institutional Animal Care and Use Committee at the Third Affiliated Hospital of Sun Yat-Sen University (ethics committee approval number: IACUC-F3-23–0615). ARRB1-KO mice were generated as previously described (Lei et al. 2021). During the experiment, mice were housed with sufficient food and water under a pathogen-free condition (12-h light/dark cycle).
For in vivo APAP-injury study, male mice (23 ± 2 g, 8 weeks old) were intraperitoneally injected with 400 mg/kg APAP (MCE cat: HY-66005), after being fasting for 12 h. Pair-fed control mice were intraperitoneally injected with a sample volume of PBS.
For inhibition of ER stress, mice were administered TUDCA (100 mg/kg body weight; MCE cat: HY-19696A) in PBS (vehicle solution) by intraperitoneal injection 2 h prior the intraperitoneal administration of 400 mg/kg APAP (Du et al. 2022; Sun et al. 2020). The mice were euthanized 12 h later using carbon dioxide inhalation as the humane method of sacrifice. For survival analysis, 21 mice were intraperitoneally injected with 750 mg/kg APAP.
Blood and liver tissue collection
Blood was collected using the eyeball blood collection method. Briefly, we compressed the mouse eyeballs to enhance their protrusion, and then, we quickly extracted the eyeballs using forceps while simultaneously collecting blood into an EP tube. The blood was incubated at 4 °C overnight and then centrifuged at 3000 r/min. The supernatants were collected for further use.
Liver tissues were collected via a midline incision. After washing with PBS, the whole liver was separated into several parts for further use. Tissue homogenization was performed using Bead Ruptor 12 (OMINI international) following the manufacturer’s instructions.
Serological analysis
Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured as previously described (Kehua Biology, Shanghai, China, 370,457, 371,368) (Lei et al. 2021).
Inflammation factor assessment
Serum IL-1β, IL-6, and TNF-α levels as well as hepatic level were measured by ELISA kit (Servicebio, GEM0003, GEM0001, GEM0004) following the instruction.
Primary hepatocyte isolation and treatment
As described previously, hepatocytes were isolated from WT and ARRB1-KO mice by 2-step collagenase perfusion. In brief, the mice were anesthetized and subjected to a 10-min perfusion with a calcium-free buffer solution. Subsequently, perfusion with 0.05% type IV collagenase (Sigma-Aldrich, St. Louis, MO, USA, cat: G5138) was performed in the portal vein. After being minced, mice livers were filtered through 70-μm filters. Afterwards, liver cells were separated by performing two rounds of centrifugation at a force of 50 g for a duration of 2 min. The isolated hepatocytes were cultured in collagen-coated plates in preparation for subsequent experiments. Cells were then incubated with or without 10 mM APAP for 12 h.
Cell culture and treatment
AML-12 cells were seeded in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM-F12) (Gibco BRL, Rockville, MD, USA, cat: C11330500BT) supplemented with 10% heat-inactivated fetal bovine serum (FBS, cat: FSP500) and 1% ITS. Cells were incubated with 5% CO2 at 37 ℃. For APAP treatment, AML-12 cells were incubated in a cultured medium containing 10 mM APAP for 24 h, with or without 1 mM TUDCA for the indicated times.
Cell viability assessment
Cell viability was evaluated according to the provided instructions (Dojindo Cell Counting Kit-8, cat: CK04). AML-12 cells were seeded in a 96-well culture (1 × 104 cells/well). Following APAP administration, cells were incubated in a cultured medium containing 10 μL of CCK-8 for 1 h at 37 °C. Cell viability was measured as optical density (OD) using 450 nm using Multiskan Spectrum (Thermo Fisher).
Plasmid transfection and interference with ARRB1
ARRB1 plasmid was bought from Umine Biotechnology Co., Ltd (Guangzhou). Plasmid transfection was performed based on the instructions of jetPRIME (Polyplus, France, cat: 101,000,046).
According to jetPRIME instructions, ARRB1 siRNA was transfected into cells for silencing. The sequence was 5′-CCUUGAGGCAUCACUGGAUAAdTdT-3′ (tsingke, China).
RNA extraction and real-time PCR for gene expression
RNA was isolated from liver tissues or cells using Trizol regent for quantitative real-time PCR (Promega, Madison, WI, USA). According to the manufacturer’s instructions, reverse transcription was performed using Reverse Transcription Kit (TOYOBO, Japan, cat: FSQ-201). SYBR Green (Invitrogen, USA) was used to assess the levels of mRNA for relevant genes with a Mini Opticon Real-Time PCR System (Bio-Rad, Hercules, CA, USA). As a normalized control, we used β-actin as a basis for determining gene expression levels. Primer sequences are listed in Table 1.
Table 1 Primer sequencesImmunohistochemistry
Mice were euthanized and sacrificed on 12 h after APAP injection. Paraffinized sections were incubated with anti-CHOP (Cell Signaling Technology, 2895) at 4 ℃ overnight. The secondary antibodies were used according to the Envision kit (DAKO, Carpinteria, CA, cat: GK500710).
The necrosis index (scoring system) for liver injury is established by Suzuki, which has 5 grades (0, no necrosis; 1, single cell necrosis; 2, necrosis < 30%; 3, 30% < necrosis < 60%; 4, necrosis > 60%) (Suzuki et al. 1993).
Immunofluorescence
Mice were euthanized and sacrificed on 12 h after APAP injection. Paraffinized sections were incubated with anti-p-eIF2α (Affinity # AF3087) used for the primary antibody. For secondary antibody, anti-rabbit Alexa Fluor 594 (Cell Signaling Technology Cat# 8889, RRID: AB_2716249) or anti-rabbit Alexa Fluor 488 (Abcam Cat# ab150077, RRID: AB_2630356) was used. The nuclear staining was performed using DAPI. A fluorescent microscope (OLYMPUS BX43) was used to photograph.
For immunocytochemistry, cells were incubated with anti-eIF2α (ZenBio Cat# 201,137) and anti-ARRB1(Abcam Cat# 32,099) at 4 ℃ overnight. Anti-rabbit Alexa Fluor 594 (Invitrogen Cat# A11037) and anti-mouse Alexa Fluor 488 (Invitrogen Cat# A11001) were used as secondary antibodies. The nuclear staining was performed using DAPI.
Protein extraction and Western blot
To extract proteins from tissue samples, 20 mg tissue was homogenized in 200 µL RIPA buffer for 40 s and lysed at 4 °C for 2 h. After centrifugation for 20 min (12,000 × g), the supernatant was collected for protein concentration analysis by the BCA method or boiled with a loading buffer for further use.
To extract proteins from cell samples, cells were lysed in RIPA buffer (100 µL/well). After centrifugation for 20 min (12,000 × g), the supernatant was collected for protein concentration analysis by the BCA method or boiled with a loading buffer for further use.
Antibodies against ARRB1 (Abcam, ab32099), ARRB2 (Sigma-Aldrich, SAB2500117), Cyp2e1 (Proteintech, 19,937–1-AP), Glucose Regulated Protein 78 (GRP78) (Cell Signaling Technology, 3177), p-eIF2α (Abclonal, AP0692), ATF4 (Proteintech, 10,835–1-AP), eIF2α (Cell Signaling Technology, 5324), CHOP (Cell Signaling Technology, 2895), cleaved caspase 3 (Affinity, AF7022), anti-JNK1 + JNK2 + JNK3 (phospho T183 + T183 + T221) (Abcam, ab124956), SAPK/JNK (Cell Signaling Technology, 9252), and GAPDH (Sigma-Aldrich, A5441), anti-Bax antibody (Abcam, ab32503), Bcl2 monoclonal antibody (Proteintech, 68,103–1-Ig), β-actin (Cell Signaling Technology, 3700), and α-tubulin (Ray antibody biotech RM2007) were used as primary antibodies.
Signals were evaluated by ECL chemiluminescence (Thermo Fisher Scientific, Waltham, MA, USA). Normalized controls included GAPDH, β-actin, and α-Tubulin. Data represents the ratio to the control group after normalization.
Liver GSH, SOD, MDA assay
GSH levels were assessed in liver tissues (20–50 mg) based on the instructions of commercially available kits (Boxbio, Beijing, cat: AKPR008M).
Superoxide dismutase (SOD) activity and malonaldehyde (MDA) levels were assessed by commercially available kits (Boxbio, Beijing, China, cat: AKAO001M, AKFA013M).
TUNEL staining
Apoptosis of samples obtained from mouse liver tissue or cells was analyzed using the deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay (Servicebio, cat: G1502). Nuclear staining was performed using DAPI. Each group includes at least 3 mice.
Flow cytometry
To detect cell death, the Annexin V-FITC/PI Apoptosis Detection Kit (BD Bioscience, US, cat: 556,547) was used to quantify the apoptosis of AML-12 24 h after incubation with 10 mM APAP. Briefly, (1 × 105 cells/well) AML-12 cells were seeded in 6-well plates. After incubation with APAP and intervention at the indicated time, cells were digested by trypsin, suspended, and incubated with reagents from the detection Kit. Flow cytometry was used to quantify the cells.
Co-immunoprecipitation (CO-IP) assay
Proteins from AML-12 were lysed in Pierce™ IP Lysis Buffer after experimental treatments. Lysates were incubated with protein A/G magnetic beads (MCE, cat: HY-K0203) at 4 °C overnight and then incubated with anti-ARRB1 (Abcam, ab32099), anti-eIF2α (ZenBio Cat# 201,137), anti-p-eIF2α (Abclonal, cat: AP0745) antibody, or IgG antibodies (Beyotime, A7016; Beyotime, A7028) overnight at 4 °C. The magnetic beads were washed with PBST and boiled in a loading buffer at 70 °C for 10 min. The supernatants were collected and subjected to Western blot.
Statistical analyses
All data were expressed as the mean ± SD. Data analysis was performed using GraphPad Prism version 8.0. A two-tailed Student’s t test or one-way analysis of variance (ANOVA) was employed to determine statistical significance. A significance level of P < 0.05 was used to determine statistical significance. Each experiment was independently replicated at least three times, and consistent results were obtained.

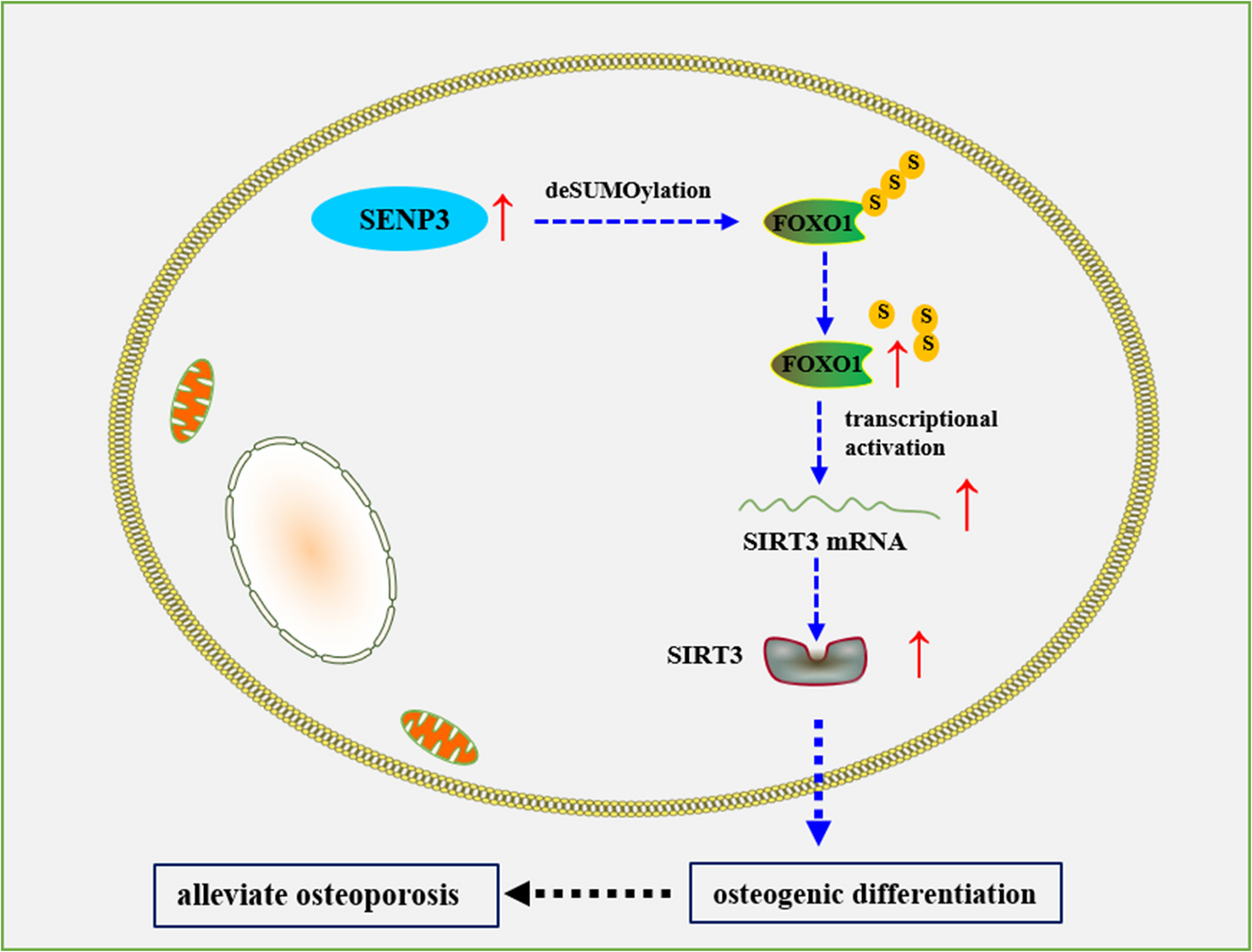
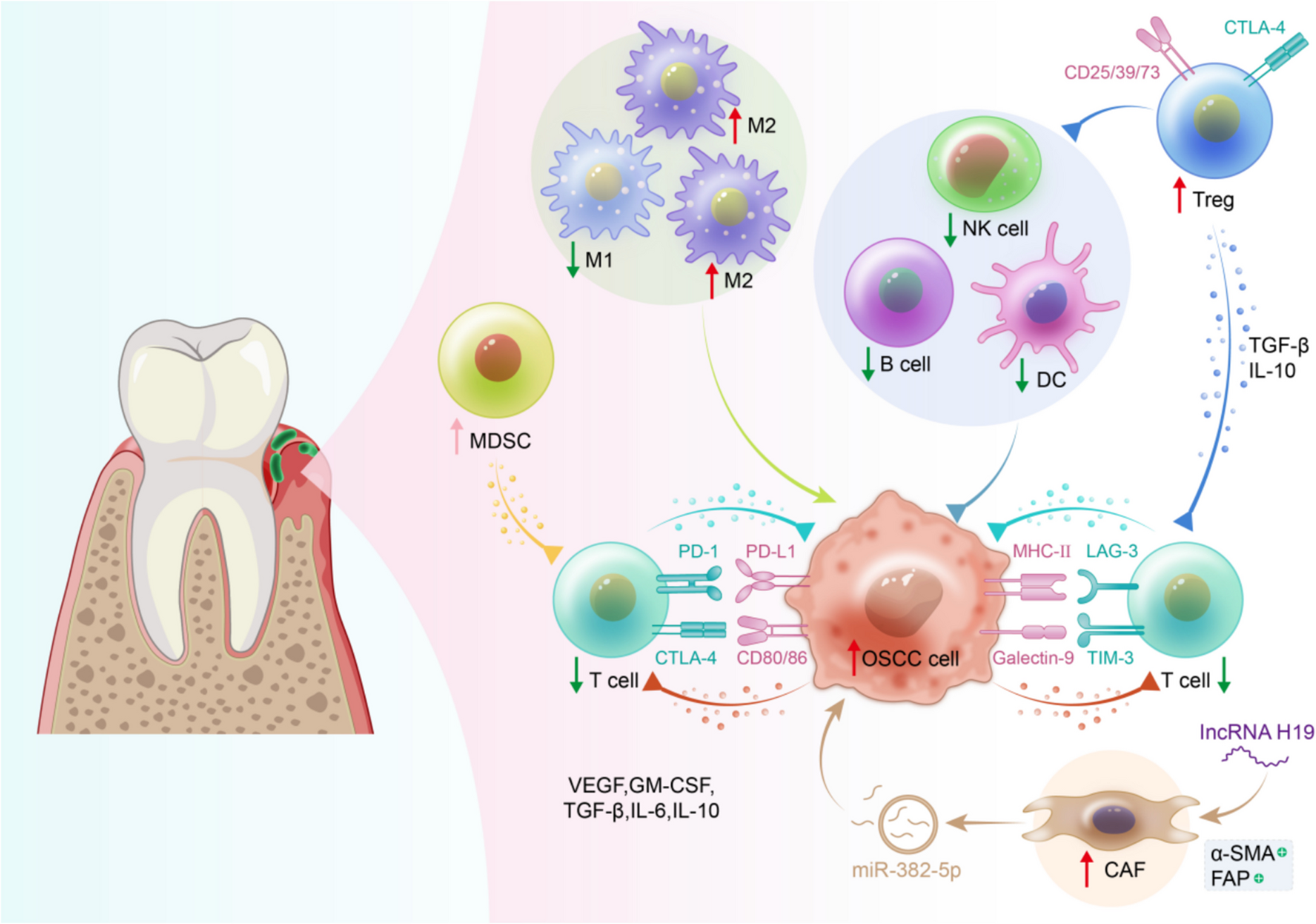
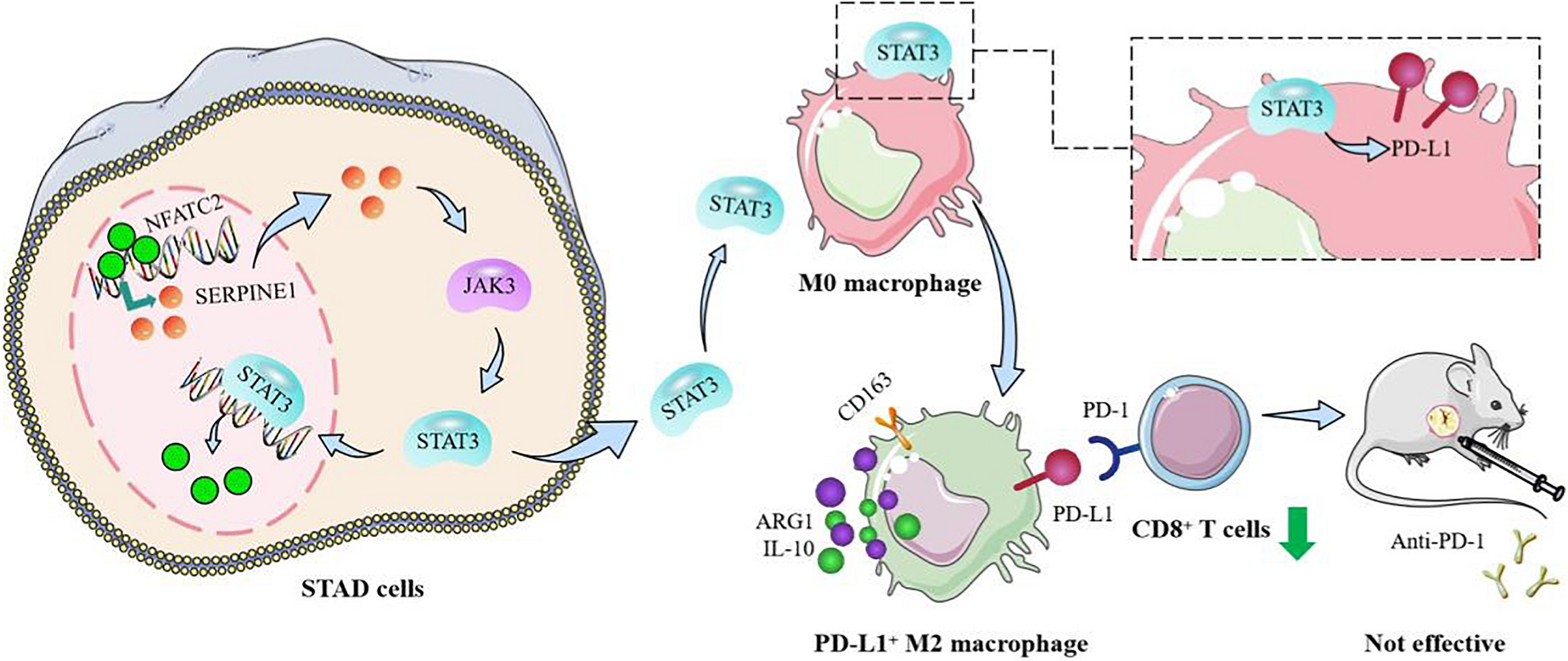
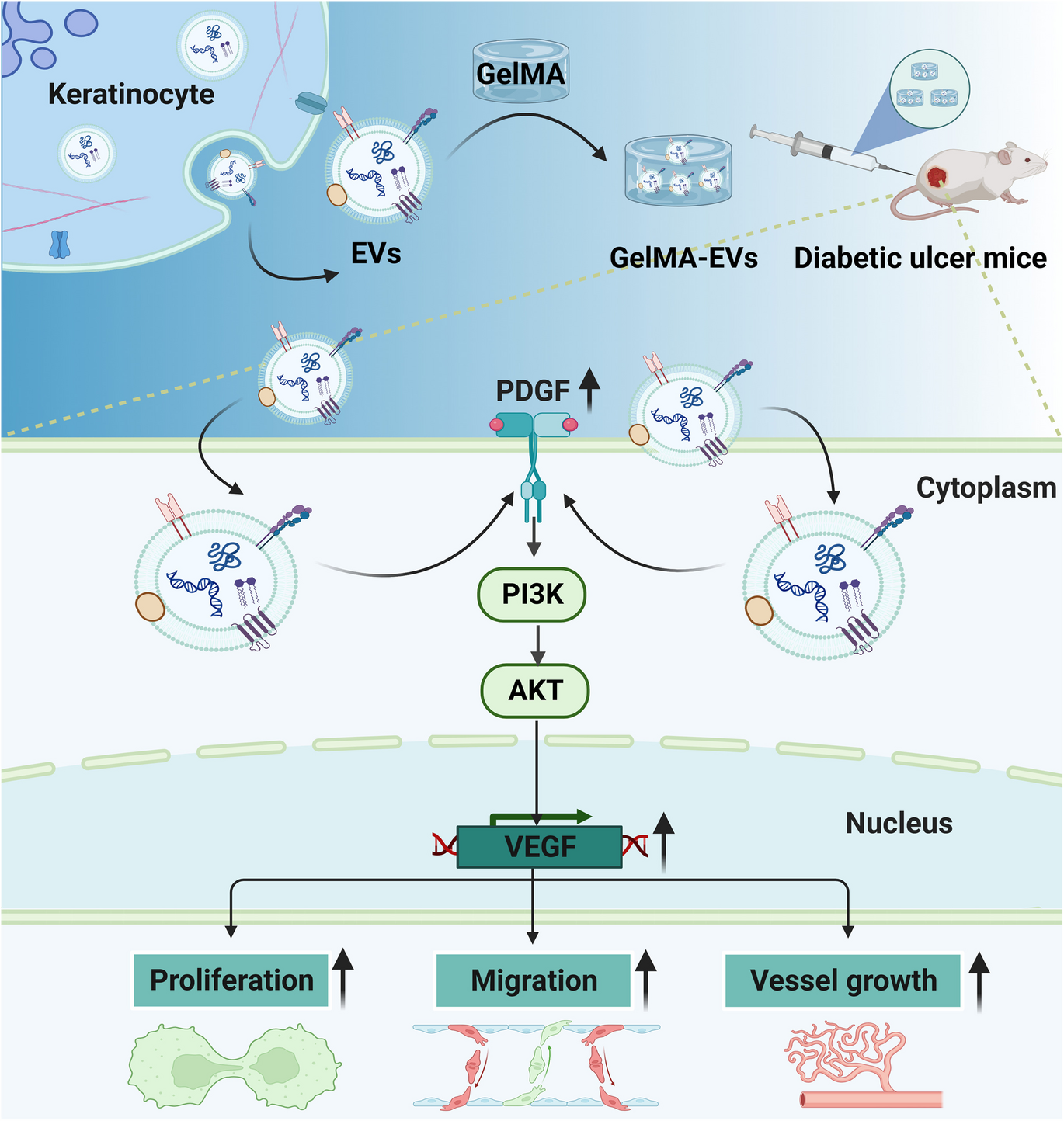
Comments (0)