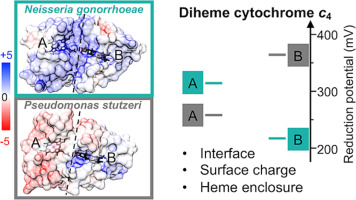
Cancer, the leading reason of death worldwide, is affected by genetic and environmental conditions and defined by uncontrolled division and proliferation of cells [1,2]. Chemotherapy is an effective cancer treatment method. Classical platinum agents, such as cisplatin, carboplatin, and oxaliplatin, are potential chemotherapeutic drugs [[3], [4], [5]] that obey the structure activity relationships (SAR) with a general formula of [PtA2X2] [6,7] (A2 = amine carrier ligand; X2 = leaving ligand) [[8], [9], [10], [11], [12], [13]]. Nuclear DNA (nDNA) is the major binding target for classical platinum agents. The anticancer activities of classical Pt(II) agents mainly result from their binding to nDNA with 1,2-GG intrastrand cross-links, causing significant DNA structural changes that inhibit DNA replication and transcription and result in cancer cell death [14,15]. However, their applications are severely limited by drug resistances and side effects [[16], [17], [18], [19], [20]]. Drug resistance is induced by the recognition and repair of proteins to the distorted Pt-DNA structures through the nucleotide excision repair (NER) pathway [21,22]. Designing drugs that damage DNA without inducing resistance via NER signaling pathway is crucial to enhance the anticancer therapy effectiveness. One way to circumvent resistance is to design novel platinum complexes that form different types of DNA cross-links. [15,[23], [24], [25]].
With a different general formula of [PtA3Cl]+, monofunctional platinum complexes, represented by pyriplatin ([Pt(NH3)2(pyridine)Cl]+) and phenanthriplatin (cis-[Pt(NH3)2(phenanthridine)Cl]+), target nDNA through a different binding mode to classical Pt(II) drugs [8,[26], [27], [28], [29]]. Since monofunctional platinum complexes contain only one leaving ligand, a single covalent bond between the Pt agents and the guanine-N7 atom in DNA is formed instead of the two bonds by classical Pt(II) agents [30]. This may help circumvent the drug resistance by initiating different downstream pathways.
Another cause of drug resistance is associate with thiol-containing biomolecules, such as glutathione (GSH), which is the second major binding target for the platinum complexes [31]. A useful strategy to prevent such resistance is introducing one or more sterically hindered ligands to the platinum center to prevent disturbance of sulfur-containing molecules prior to the binding to DNA [[32], [33], [34]]. In the pyriplatin and phenanthriplatin, as the steric hinderance of planar heterocyclic carrier ligands increase, higher anticancer activity is achieved [8,26,35]. Coincidently, molecular dynamic simulation also revealed that larger conformational distortions of DNA are induced by phenanthriplatin with more hindered carrier ligands [36]. Some studies also suggest that larger DNA conformational changes including unwinding/bending are caused by larger steric hindrance of carrier ligands to accommodate the Pt(II) agents in the DNA double helix [37,38]. We thus suspect that the monofunctional Pt(II) complexes with larger steric hindrance may further induce larger DNA structural alterations and show higher anticancer activities.
To minimize the side effects of classic platinum anticancer drugs, cancer-targeted platinum agents have been developed [[39], [40], [41], [42], [43], [44]]. In addition to nDNA, mitochondrial DNA (mtDNA) is also the binding target for cancer cells, the damage of which can elude DNA self-repair and cause death of cancer cells [[45], [46], [47]]. Triphenylphosphonium (Ph3P+, TPP), as a mitochondrial targeting group, could locate platinum agent in mitochondria [48,49]. Recently, three monofunctional Pt(II) complexes (namely OPT, MPT and PPT) were designed and synthesized [50], with the –CH2Ph3P+ group at ortho, meta and para positions of the pyriplatin pyridine ring. Quantum mechanical computationally study on OPT, MPT and PPT were used to investigate their DNA binding ability and potential interactions with non-DNA targets, and so on [51]. Among the three complexes, OPT is the most concentrated complex found in mitochondria in vitro experiments, leading to the damaged mtDNA and higher activity against non-small-cell lung cancer [50]. So in this study, modification of the ligands are based on OPT rather than the other two complexes. The sterically hindered ligand (ortho-PPh3CH2Py) in OPT [20] (Fig. 1a) slows down the reactions of GSH and OPT, reducing drug resistance caused by GSH [[32], [33], [34]]. As mentioned above, the monofunctional Pt(II) agents with larger steric hindrance may induce larger DNA distortions and show higher anticancer activities. Bearing these in mind, we introduced 1,10-phenanthroline (phen) and 4,7-diphenyl-1,10-phenanthroline (4,7-Ph2phen) to replace the two NH3 ligands in OPT as carrier ligands, which introduce larger steric hindrance compared to the NH3 ligand [40,41,52] with 4,7-Ph2Phen group being the most hindered. They are named OPT-II and OPT-III, respectively (Fig. 1b and c).
Because DNA structural distortions in the Pt-DNA adducts caused by TPP-modified monofunctional platinum complexes are not yet clear but could be helpful in explaining the antitumor activity of these platinum drugs (a hot topic on its own), we constructed their Pt-DNA adducts by combined OPT, OPT-II, OPT-III to the dodecamer DNA duplex 5′-d(C1C2T3C4T5C6G*7T8C9T10C11C12)-3′·5′-d(G1G2A3G4A5C6G7A8G9A10G11G12)-3′ (G* indicates the binding site of the OPT, OPT-II, OPT-III agents) based on the X-ray structure of the pyriplatin-DNA adduct [8] with a standard B-DNA duplex as reference (Fig. 1d). Molecular dynamic (MD) simulations for the OPT-DNA, OPT-II-DNA, OPT-III-DNA adducts and undamaged DNA were performed to obtain detail information of DNA structural changes with different carrier ligands.


Comments (0)