Materials
Dulbecco’s phosphate buffered saline (PBS), fetal bovine serum (FBS), Pierce BCA Protein Assay kit, Griess Reagent Kit, Macrophage-SFM (1X) media, Pierce 20x TBS buffer, mouse TNF-α uncoated ELISA kit, and mouse IL-6 uncoated ELISA kit were all purchased from Thermo Fisher Scientific (Grand Island, NY). Dulbecco’s Modified Eagle’s Medium F-12 Ham (DMEM/F-12), trypsin, trypan blue, bromophenol blue, β-mercaptoethanol, dimethyl sulfoxide (DMSO), bovine serum albumin (BSA), sodium hydroxide (NaOH), Tween-20, thiazolyl blue tetrazolium bromide (MTT), penicillin/streptomycin, Roche cOmpleteTM Protease inhibitor cocktail tablets, PhosSTOP tablets, LPS from Escherichia coli O111:B4, and 2′,7′-dichlorofluorescin diacetate (DCF-DA) were all purchased from Sigma-Aldrich (St Louis, MO). MACS® CD11b+ Mouse MicroBeads, MS Columns, and the Adult Mouse Brain Dissociation Kit were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany). iTaq Universal Probes One-Step kit (containing 2x probes RT-PCR reaction mix, iScript reverse transcriptase, and nuclease-free water), 0.2 µm nitrocellulose membrane, 0.45 µm immuno-blot low fluorescence PVDF membrane, Precision Plus Stained protein ladder, Mini-PROTEAN TGX 4-15% and 4-20% precast gels, and extra thick blotting paper were purchased from BioRad (Hercules, CA). QIAshredder columns, RNeasy Mini Plus kit, HiPerfect transfection reagent, FlexiTube FABP4 siRNA (SI02695322), and AllStar Negative Control siRNA were purchased from Qiagen (Hilden, Germany). LI-COR Intercept® blocking buffer, LI-COR donkey anti-rabbit antibody (680 nm), and primary β-actin antibody were purchased from Millennium Science (Melbourne, Victoria, Australia). Anti-FABP3 antibody (ab45966) and anti-FABP4 antibody (ab66682) were obtained from Abcam (Cambridge, United Kingdom). Anti-FABP5 antibody (39926), anti-UCP2 antibody (D105V), SAPK/JNK antibody (9252), and phospho-SAPK/JNK (Thr183/Tyr185) antibody (81E11) were purchased from Cell Signalling Technology (Danvers, MA). Anti-TLR4 (48-2300) antibody was purchased from Thermo Fisher Scientific and BMS309403 was purchased from Cayman Chemicals (Ann Arbor, MI). 3H-oleic acid (3H-OA) was purchased from American Radiolabelled Chemicals Inc (St. Louis, MO), while 3H-2-deoxy-D-glucose (3H-2-DG) and Ultima Gold liquid scintillation cocktail were purchased from Perkin-Elmer (Boston, MA).
Culturing of BV-2 Cells and Primary Mouse Microglia
BV-2 cells, originating from the laboratory of Dr. Elisabetta Blasi (Blasi et al. 1990), were generously provided by Dr. Linda J. Van Eldik (Lexington, KY). BV-2 cells were cultured in DMEM/F-12 media supplemented with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin. Cells were seeded into required plates at 20,000 cells/cm2, which were left in the 37 °C, 5% CO2 incubator until they reached 80% confluency and were used for experiments.
All experiments requiring the use of mice for isolation of microglia were approved by Monash Institute of Pharmaceutical Sciences Animal Ethics Committee (MIPS.27467) and performed in accordance with the National Health and Medical Research Council of Australia guidelines for the care and use of animals for scientific purposes. Brains removed from anesthetised C57BL/6 female mice (6-8 weeks old) were digested into a single cell suspension using the Adult Brain Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) and Miltenyi Biotec gentleMACS™ Dissociator as per the manufacturer’s protocol. CD11b+ cells were then magnetically selected using the MACS® MicroBeads and MS Columns (Miltenyi Biotec). The selected cells were seeded at 50,000/cm2 into required plates and maintained in Macrophage-SFM media containing 1% (v/v) penicillin/streptomycin in a 37 °C, 5% CO2 incubator until confluent.
Cell Treatment
The day after seeding, BV-2 cells were treated with 1 µg/mL LPS for 24 h. For FABP siRNA silencing, BV-2 cells were transfected 4 h post seeding as per the protocol previously described (Low et al. 2021). In brief, BV-2 cells were transfected with either FABP siRNA or AllStar Negative Control (siCont) for 5 min. After that, the concentration of siRNA complexes was diluted to 5 nM with FBS-containing media and cells were incubated with the siRNA treatment for 24 h prior to LPS being added to the cells. For BMS309403 treatment, BV-2 cells were co-treated with LPS (1 µg/mL) and BMS309403 (50 µM in vehicle) or vehicle (0.1% (v/v) DMSO) 24 h post-seeding and incubated for 24 h prior to being used for experiments. To assess the impact of BMS309403 post-microglial activation, BMS309403 (50 µM in vehicle) or vehicle (0.1% (v/v) DMSO) was added to BV-2 cells that had already been treated with LPS (1 µg/mL) for 4 h. To assess the impact of FABP4 inhibition in primary mouse microglia, 50 µM BMS309403 or vehicle (0.1% (v/v) DMSO) was added for 2 h prior to the addition of LPS (0.1 µg/mL) and microglia were co-treated for 24 h prior to being used for experiments. To assess the phosphorylation activity of JNK and total level of JNK, BV-2 cells were treated with LPS (1 µg/mL) for 30 min with or without siRNA or BMS309403 treatments.
Western Blotting (WB) for Quantifying Protein Expression of FABPs, TLR4, UCP2, and P-JNK/JNK in BV-2 Cells
Following treatment, protein levels of FABPs, TLR4, UCP2, and p-JNK/JNK in BV-2 cells were quantitatively assessed using WB. Post-treatment, BV-2 cells were washed with ice-cold PBS prior to being lysed using Pierce IP lysis buffer supplemented with Roche cOmpleteTM mini protease inhibitor. Following centrifugation at 13400 xg for 10 min at 4 °C, protein count was determined using the Pierce BCA Protein Assay kit. 10 μg of protein was loaded in a 5:1 ratio with 6x Laemmli loading buffer on a BioRad 4-20% acrylamide precast gel, where the gel was run at 200 volts for 55 min using a BioRad Mini-PROTEAN Tetra cell (Hercules, CA). Following electrophoresis, the gel, alongside two thick blotting papers and a 0.2 μm nitrocellulose membrane, was equilibrated in transfer buffer containing 20% (v/v) methanol for 30 min. The protein from the gel was then transferred onto the nitrocellulose membrane at 11 volts for 42 min using a BioRad semi-dry transblot (Hercules, CA). The membrane was incubated in LI-COR Intercept® blocking buffer before being incubated overnight at 4 °C with primary antibodies to specific mouse FABP isoforms (1:5000 dilution), TLR4 (1:500 dilution) or UCP2 (1:1000 dilution) and anti-mouse primary β-actin (1:10000 dilution) diluted in LI-COR Intercept® blocking buffer. The membrane was further incubated in donkey anti-rabbit secondary antibody (1:30000 dilution) for 2 h at room temperature. The membrane was imaged using an AmershamTM Typhoon scanner (Marlborough, MA). Densitometric analysis on WB bands was performed using Image J (Bethesda, MD), with the proteins of interest quantified relative to β-actin (housekeeping protein).
Proliferation Assay and Cell Viability
The proliferation rate of BV-2 cells prepared in 3 different plates was investigated by simply counting the number of cells on a haemocytometer before and after LPS treatment for 12 and 24 hours. To ensure that the concentrations of BMS309403 used on BV-2 cells did not significantly affect cell viability, an MTT assay was performed. Post-treatment, BV-2 cells were washed once with PBS prior to adding 150 µL of 0.45 mg/mL MTT reagent (prepared in FBS-free DMEM/F-12 media) to each well. The plate was incubated at 37 °C with 5% CO2 for 4 h. After incubation, the MTT reagent was removed and 150 µL of DMSO was added into all wells for 30 min. The plate was immediately read on a Perkin-Elmer Enspire fluorescence plate reader (Boston, MA) at 540 nm. Following background subtraction, the percentage cell viability was expressed as a ratio of mean absorbance of BMS309403-treated cells over mean absorbance of vehicle-treated cells.
Quantification of 3H-OA Oxidation Rate in BV-2 Cells
The oxidation rate of 3H-OA in BV-2 cells was measured as reported previously (Ma et al. 2020). Briefly, BV-2 cells were incubated with incubation medium containing 60 µM of OA and 1 μCi 3H-OA for 4 h at 37 °C with 5% CO2. Following incubation, a 100 μL aliquot of supernatant was removed and mixed with an equal volume of ice-cold 10% (v/v) trichloroacetic acid. The mixture was centrifuged at 13,000 xg for 5 min at 4 °C. A 150 μL aliquot of the resulting supernatant was then treated with cold methanol:chloroform (2:1) and 2 M KCl:HCl before being centrifuged at 3,000 xg for 5 min. An aliquot of the upper (aqueous) phase (containing the oxidised 3H-OA, 3H2O) was removed and mixed thoroughly with 2 mL of scintillation fluid (Ultima Gold cocktail) and the radioactivity was determined using a Perkin-Elmer 2800TR liquid scintillation counter (Boston, MA). Oxidised 3H-OA (3H2O) produced was normalised to total protein count (μg) using a BCA assay and reported as pmol/μg protein.
Cellular Uptake of 3H-2-DG
BV-2 cells and primary mouse microglia were rinsed with blank media before being supplemented with 1 μCi of 3H-2-DG prepared in culture media. Uptake was ceased after 15 min by removing the 3H-2-DG and rinsing the cells with ice-cold PBS three times. The cells underwent a freeze-thaw cycle before being lysed using Pierce IP lysis buffer and mixed thoroughly with 2 mL of scintillation fluid (Ultima Gold cocktail) and the radioactivity was determined. The amount of 3H-2-DG in cells was normalised to total protein count (μg) using a BCA assay and reported as pmol/μg protein.
Measurement of Intracellular ROS and NO Production
For the measurement of intracellular ROS, BV-2 cells were rinsed once with warm HBSS and then incubated with 40 µM of DCF-DA prepared in FBS-free media for 30 min at 37 °C in 5% CO2. Following incubation, the wells were rinsed once with warm HBSS and HBSS was added back into the wells before being read on a Perkin-Elmer Enspire plate reader (Perkin-Elmer) (λex: 488 nm, λem: 535 nm).
For measurement of NO production, BV-2 cells were aliquoted into a 96-well plate, and the Griess assay was performed as per the manufacturer’s protocol (Molecular Probes, Eugene, OR). The plate was then read on a Perkin-Elmer Enspire plate reader at 548 nm. Nitrite concentrations in samples were back calculated from a standard curve prepared through serial dilution of NaNO2 prepared in media.
Quantification of Proinflammatory Cytokine Levels Using ELISA
The ELISAs were performed as per the manufacturer’s protocol to measure the amount of proinflammatory cytokines (TNF-α and IL-6) released from BV-2 cells and primary mouse microglia following FABP4 knockdown and inhibition, in the presence and absence of LPS. Post-treatment, 100 μL of supernatant, alongside either 100 μL of TNF-α or IL-6 standards, was aliquoted into pre-coated ELISA plates. Following a 2 h incubation, the wells were washed with wash buffer (0.05% (v/v) Tween-20 in PBS) four times before 100 μL of detection antibody (specific to the cytokine of interest) was added into each well for 1 h. The wells were then washed four times with wash buffer before 100 μL of Streptavidin-HRP was added into each well for 30 min. The wells were again washed six times before the wells were incubated with 100 μL of TMB substrate for 15 min. Following the addition of 100 μL of Stop solution, the plates were immediately read on a Perkin-Elmer Enspire fluorescence plate reader at 450 nm and 570 nm. Absorbance values at 570 nm were subtracted from the values at 450 nm to account for background. The concentration of cytokines released was normalised to total protein count (mg) determined with a BCA assay and reported as pg/mg protein.
Data and Statistical Analyses
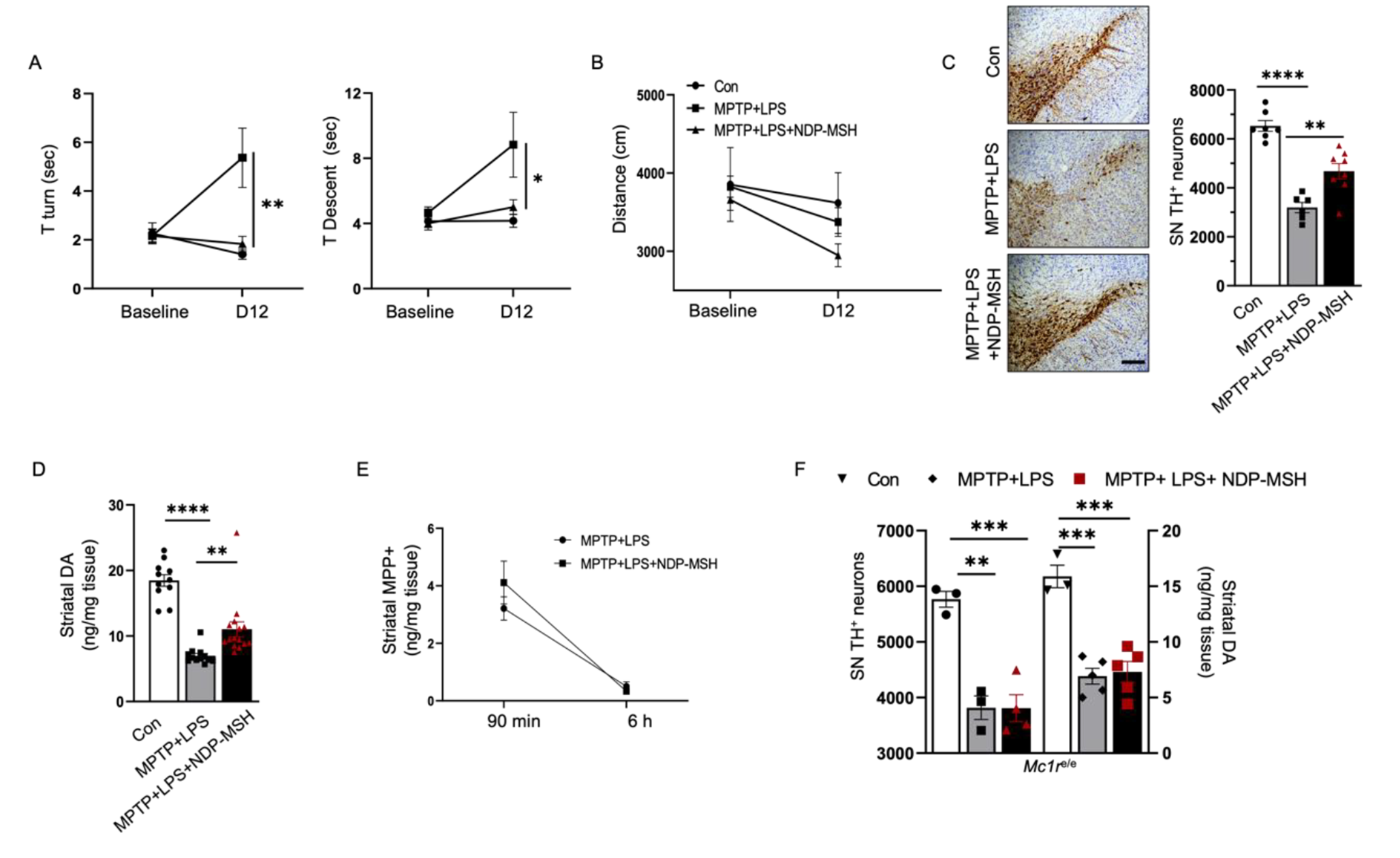
Data analyses were performed using Graphpad Prism 8. All data were expressed as mean ± SEM, where all replicates performed were biological replicates. When comparing between two groups, a Student’s unpaired t-test was performed. When more than two groups were compared, an analysis of variance (ANOVA), followed by an appropriate post-hoc test, as defined in the figure legends, was performed. Values of p<0.05 were considered statistically significant.




Comments (0)