
Remember me



Low-dose naltrexone (LDN), in daily doses of 1 to 5 mg,45 is, due to its potential analgesic and anti-inflammatory effects, increasingly used as an off-label treatment of fibromyalgia (FM) and some autoimmune pain conditions.10,36,37,45,57 Naltrexone is well-known for its use in the treatment of opioid and alcohol addiction in daily doses of at least 50 mg. Naltrexone is a µ-opioid-receptor antagonist with, to a lesser extent, δ-receptor antagonistic properties, bearing a close structural similarity to naloxone. Increased oral bioavailability and longer half-life (T1/2β) for the active metabolite 6β-naltrexol makes naltrexone pharmacologically preferable over naloxone.57 Recent experimental studies have demonstrated that LDN acts as an immune modulator in the CNS.19,31Low-dose naltrexone has been used in disorders with a putative significant neuroinflammatory component, eg, chronic pelvic pain, CRPS (complex regional pain syndrome), interstitial cystitis, epilepsy, FM, inflammatory bowel disease, and multiple sclerosis.24,32,57
Fibromyalgia is a chronic, nociplastic,2,4 musculoskeletal disease with an unknown etiology characterized by widespread fluctuating pain, fatigue, low quality of sleep, and high incidences of depression and anxiety disorders.23,43 In Europe and the United States, 2% to 8% of the population experience FM,12 affecting women more frequently than men.7,55 The main drug classes recommended in FM are antidepressants, anticonvulsants, and opioids.2,30 Although demonstrating clear evidence for analgesic efficacy, these drugs are associated with a risk of initiating severe arrhythmias, cardiac dysfunction, neuropsychiatric disorders, serotonergic syndrome, and enhanced postoperative morbidity.18 Anticonvulsants have been recommended as alternatives associated with the development of dependence, substance abuse, and suicidality.27,33 European League Against Rheumatism (EULAR) recommends that tramadol can be used in severe pain when nonpharmacological multimodal therapies fail.25 Paradoxically, in clinical FM studies, opioids generally demonstrate limited analgesic efficacy,15 but because tramadol also has serotonin-norepinephrine reuptake Inhibitor (SNRI) properties, this may explain the observed weak analgesic effect. All opioids, including tramadol,40 carry a risk for the development of tolerance, dependence, and substance abuse, highlighted by the “opioid epidemic” in the United States.30,47
Small-scale studies10,55,56 indicate analgesic efficacy of LDN with few adverse effects, and thus, our rationale for this study was to corroborate the findings in a higher volume.
Only 6 studies address LDN as a treatment of FM. For example, a pilot study by Younger et al. described lowered symptoms in all included 10 patients with FM when receiving LDN.55 A randomized, double-blind, placebo-controlled study also by Younger et al.56 showed a significant decrease in pain intensity in 31 included patients with FM. A study investigating the dose–response relationship among 25 patients with FM10 found that 4.5 mg/day was effective in 95%. A study including 8 patients with FM showed a decrease in proinflammatory cytokines and less pain and symptoms after 8 weeks of LDN treatment.31 An explorative study describing cold pressor test (CPT) to measure pain included 15 patients with FM and showed improved scores after receiving LDN.29 Finally, an explorative study using CPT in patients with chronic opioid treatment showed improved CPT scores among 21 patients with FM after LDN treatment.22
The main objectives of this study were as follows: First, to examine if LDN was associated with a higher analgesic efficacy and improvement in physical function score compared with placebo. Second, to ascertain the analgesic efficacy of LDN in experimental pain procedures using quantitative somatosensory testing.9,28Third, to examine the pharmacokinetics of LDN and the main metabolite 6β-naltrexol.
2. Methods 2.1. Study managementThe Committee of Health Research Ethics of The Region of Southern Denmark (S-20150159), the Danish Medicines Agency (2015102044), and the Data Inspection Authority of The Region of Southern Denmark (2008-58-0035) approved the study protocol. The study was registered in EUDRACT (2015-002972-26) and ClinicalTrials.gov (NCT02806440) and conducted in accordance with Good Clinical Practice (GCP) and Good Manufacturing Practice (GMP).
2.2. Investigational centersThe study was planned as a 2-center collaboration between The Multidisciplinary Pain Center at Rigshospitalet, Copenhagen (MPC-C), a tertiary university facility, and The Multidisciplinary Pain Clinic at Friklinikken, Grindsted, (MPC-G), a secondary health care facility. In total, an enrollment of 140 patients with FM was planned, with 70 patients with FM allocated at each investigational center. Due to organizational changes at MPC-C, only 1 patient was randomized at MPC-C. This patient made a withdrawal of consent after intake of 1 tablet. The study continued as 1 investigational center, MPC-G, planned to include 70 patients with FM.
2.3. Study design 2.3.1. Study settingThe study was an investigator-initiated and investigator-driven study using a block-randomized, double-blind, placebo-controlled, crossover design (see Fig. 1 showing overview of study). The patients with FM were randomized to treatment with LDN or placebo in the first or second treatment period. The patients with FM were randomized and allocated equally according to computer random table method. The pharmacy managed the randomization sequence generated by a web-based randomization site.4 The sequence was generated using the second generator function, applying blocks of 10 and balanced permutations (see text, Supplemental Digital Content 1, text describing study setting, available at https://links.lww.com/PR9/A195).
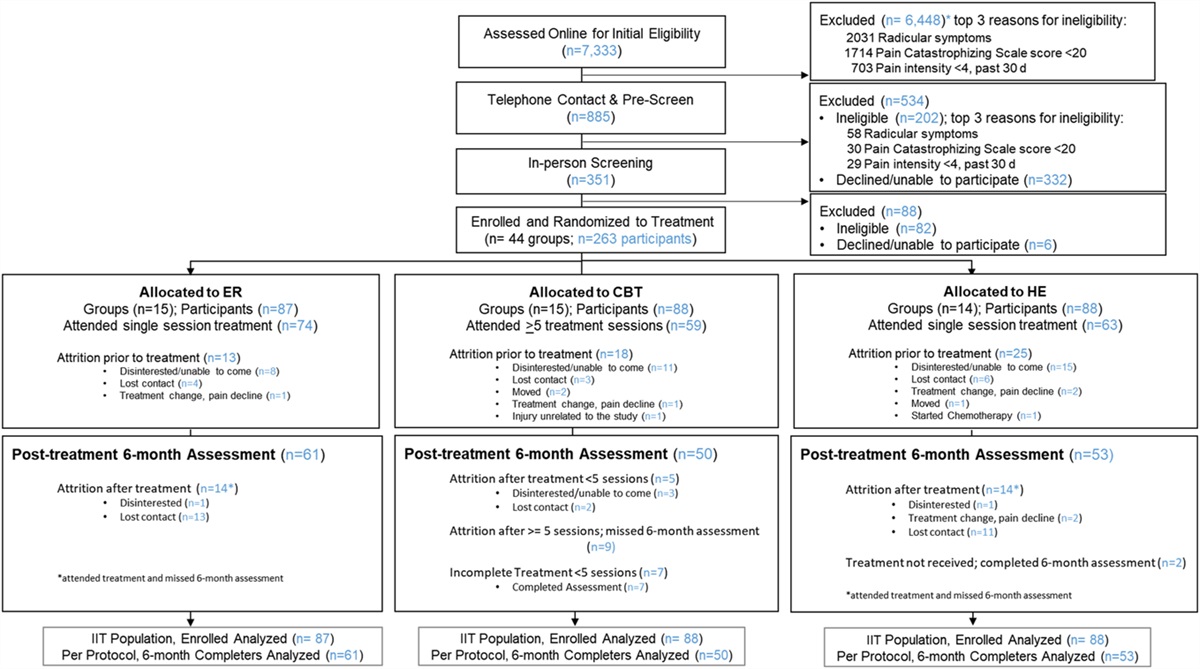
 Figure 1.:
Figure 1.: Consort (2010) flow diagram, patient selection, and eligibility. LDN, low-dose naltrexone.
The randomization lists were stored in secure and locked confines, only accessible by the principal investigator. In case of a medical emergency, the code could be individually unblinded. Patients with FM were included consecutively and evenly over time. New patients with new randomization numbers were allocated to replace dropouts. Patients, study staff, and staff in pain clinic were blinded throughout the study period. The study was executed transparently and presented in accordance with CONSORT guidelines. Information and data collection from patients were done by 5 trained staff members. Study data were manually entered into an electronic case report form (e-CRF). After the last patient visit, data were stored in OPEN, a dedicated research registry in The Region of Southern Denmark (see text, Supplemental Digital Content 1, text describing study setting and source data, available at https://links.lww.com/PR9/A195).
2.4. Outcomes 2.4.1. Primary outcomesThe primary outcomes were patients with FM reporting on function, total impact, and symptoms in Fibromyalgia Impact Questionnaire revised (FIQR)5 questionnaire (cf. 2.8.2.1) and reporting on pain intensity using the summed pain intensity ratings (SPIR)8,26 (cf. 2.8.2.2).
2.4.2. Secondary outcomesThe secondary outcomes were as follows:
(1) Diary-based questionnaires; Brief Pain Inventory-Short Form (BPI-SF),51 Daily Sleep Interference Scale (DSIS),48 Hospital Anxiety and Depression Scale (HADS),58 PainDETECT Questionnaire (PD-Q),16 and Pain Catastrophizing Scale (PCS)35,42 (cf. 2.8.2.3) (2) Quantitative Somatosensory Testing (QST)28 paradigms (cf. 2.9) (3) Pharmacokinetics of naltrexone and the main metabolite 6β-naltrexol (cf. 2.10.2) 2.4.3. TimelineThe study had a 3-phase setup (see Fig. 2 showing study phases). The first phase included baseline assessment (BA1) (day −3 to day 1) and a treatment period (day 1 to day 21) including an outcome assessment (OA1). The second phase was a washout period (day 22 to day 32). The third phase included a baseline assessment (BA2) (day 33 to day 36) followed by a treatment period (day 36 to day 56) including an outcome assessment (OA2). The patients with FM attended 6 separate examination days (see Table 1, showing overview of the study).
 Figure 2.:
Figure 2.: Study design. Patients were randomized and allocated in a double-blind design to follow (A) or (B). The 2 treatment periods were identical (21 days) and separated by a washout period (14 ± 2 days). Before each treatment period, baseline assessments (questionnaires, experimental pain testing, and blood samples) were performed, providing a clinical status of the patient.
Table 1 - Overview of the study showing the 3 phases including 2 treatment periods separated by the washout period.
The first and second baseline assessment (BA1/BA2) and the first and second outcome assessment (OA 1/OA 2) are shown. Detailed day-by-day timetable for the completion of examination days, study questionnaires, pain ratings, blood sampling, quantitative sensory testing, diary, and medication are shown.
BA1, baseline assessment period 1; BA2, baseline assessment period 2; BPI, Brief Pain Inventory - Short Form; CPM, conditioned pain modulation; Diary, days for completion the diary; DSIS, Daily Sleep Interference Scale; FIQR, Fibromyalgia Impact Questionnaire revised; HADS, Hospital Anxiety and Depression Scale; Medication, days with consumption of project medication LDN/Placebo; OA1, outcome assessments period 1; OA2, outcome assessments period 2; PCS, pain catastrophizing scale; PPT, pain pressure threshold; PD-Q, PainDetect Questionnaire; QST Hyperalgesia + Allodynia, heat/capsaicin test; SPIR, summed pain intensity rating.
Naltrexone (4.5 mg) and identical placebo tablets were manufactured and packed in a blinded and randomized fashion by the pharmacy (magistral production, Glostrup Apotek, Copenhagen, Denmark). Naltrexone (4.5 mg) is not marketed in Denmark but manufactured by permission of the Danish Medicines Agency (DMA), controlling the authorization and licensing of the manufacturing process according to GMP. Naltrexone 4.5 mg was chosen for this study because it is the typically applied dosage in existing studies.39,56
2.6. PatientsAll patients with FM were screened by a medical specialist in rheumatology and fulfilled the ACR's (American College of Rheumatology) 2011 criteria of FM3,52–54 before enrollment in the study. The patients with FM were recruited from the patient registries at MPC-G.
2.6.1. Inclusion and exclusion criteriaInclusion and Exclusion criteria are indicated in Table 2 (see Table 2, showing inclusion and exclusion criteria).
Table 2 - Inclusion and exclusion criteria. Inclusion criteria ≥18 y of age Diagnosed with fibromyalgia according to the criteria of ACR* by a rheumatologist Premenopausal women in contraceptive treatment† or sterilized Patient referred to multidisciplinary pain treatment Exclusion criteria Other inflammatory rheumatologic diseases Pregnant/lactating Opioid treatment Cancer diagnosis Unstable analgesic medication‡ Non-proficient in Danish or English Allergy towards opioids Severe hepatic insufficiency Severe renal insufficiency Acute pancreatitis Patients were withdrawn from the study if the investigator/sub-investigator deemed it necessary due to medical reasons.*American College of Rheumatology (2011 Wolfe F, Häuser W. Fibromyalgia diagnosis and diagnostic criteria. Ann Med 2011; 43:495–502).
†Contraceptives defined as spiral or hormonal contraceptive drugs (birth control tablets, implants, transdermal depot patches, vaginal ring, or injectable depot).
‡Paracetamol rescue is accepted (requires registration in the diary).
Concomitant medications were registered in the e-CRF, including generic names and doses. Paracetamol was used as rescue medication (1 g maximum 3 times a day).
2.8. Study chronology 2.8.1. DiaryThe patients with FM received a diary that allowed entries concerning study medication, adverse events, pain assessment forms, questionnaires, and adverse events.
2.8.2. QuestionnairesThe questionnaires were self-reported. Patients were contacted by phone the day before answering the first questionnaire.
2.8.2.1. Fibromyalgia impact questionnaire revisedThe FIQR5 is a tool developed to assess FM-related problems and response to a given treatment. The FIQR was translated from the original English version to Danish by 4 health care professionals specialized in the management of chronic pain. The back-translation was then performed by a native English individual fluent in Danish. After revision and back-translation, a final revised Danish version of FIQR was generated. The FIQR explores 3 domains: function, total impact, and symptoms. The patient was asked to answer based on the experience during the last 7 days before filling in the questionnaire. The questionnaire includes 21 questions regarding everyday activities. The patient was asked to mark the degree of difficulty spanning from “no difficulty” to “very difficult performing the activity.” Furthermore, the questionnaire evaluated whether the patient was restricted or incapacitated in doing the weekly chores by the FM symptoms. The FIQR also assessed current pain intensity, energy level, sleep quality, anxiety symptoms, feeling depressed, body stiffness, sensitivity to touch, difficulties with balance, memory, and with the perception of loud, shrill noises, smells, or cold.
The FIQR scoring was made by dividing the function domain sum (0–90 points) by 3. The overall impact domain was left unchanged (0–20 points). The symptom domain sum (0–100 points) was divided by 2. The 3 domain scores were then summed (0–100 points). Differences in mean score in FIQR between BA1 (day −3 and day 1) and BA2 (day 33 and day 36) and between OA1 (day 18 and day 21) and OA2 (day 53 and day 56), respectively, were calculated.
2.8.2.2. Numeric rating scale, summed pain intensity ratingThe 11-point numeric rating scale (NRS)8,26 was applied to evaluate pain intensity (during rest, personal hygiene measures, and activity of daily living). The patient with FM indicated the pain intensity (0–10; 0 = “no pain”; 10 = “worst possible pain”) based on the experience during the last 24 hours before filling in the questionnaire. The NRS ratings across each activity were summed as SPIR (0–30 points). Differences in mean score in SPIR between BA1 (days −2, −1, 1) and BA2 (days 34, 35, 36) and between OA1 (days 19, 20, 21) and OA2 (days 54, 55, 56), respectively, were calculated.
2.8.2.3. Miscellaneous questionnairesThe BPI-SF,51 DSIS,48 HADS,58 PD-Q,16 and PCS42 are described in detail in Supplemental Digital Content (see text, Supplemental Digital Content 2, text describing miscellaneous questionnaires, available at https://links.lww.com/PR9/A195).
2.9. Quantitative somatosensory testingQuantitative somatosensory testing (QST) is a standardized activation of the sensory system by the application of graded chemical, electrical, mechanical, or thermal test stimuli, with an assessment of the evoked psychophysical responses, examining sensory detection and pain thresholds.28
2.9.1. Heat–capsaicin sensitizationThe heat–capsaicin sensitization test is a validated experimental pain model investigating aspects of central sensitization, eg, secondary hyperalgesia and allodynia13,34,38 (see text, Supplemental Digital Content 3, text describing quantitative somatosensory testing, available at https://links.lww.com/PR9/A195).
2.9.2. Pressure pain thresholdsAssessments of pressure pain thresholds (PPTs) were performed with a calibrated pressure algometer34 (see text, Supplemental Digital Content 3, text describing quantitative somatosensory testing, available at https://links.lww.com/PR9/A195).
2.9.3. Conditioned pain modulation testThe conditioned pain modulation (CPM) test evaluates the efficiency of the descending inhibitory pathways and has been used as a quantitative measure of pain disinhibition in patients with FM.9 The CPM test was performed as a cold pressor test; PPT1 was the baseline assessment, and PPT2 the assessment measured after the patient had submerged their left hand in cold water for 60 seconds (see text, Supplemental Digital Content 3, text describing quantitative somatosensory testing, available at https://links.lww.com/PR9/A195).
The CPM efficiency was calculated as follows:CPM efficiency (%)=100%×(PPT2−PPT1)PPT1.
2.10. Blood Sampling 2.10.1. Routine blood chemistryScreening of kidney and liver function was tested before inclusion in the study according to safety criteria.
2.10.2. Naltrexone and 6β-naltrexol plasma concentration measurementsTo examine the pharmacokinetics (PK) of naltrexone and its main metabolite, 6β-naltrexol, venous blood samples were collected in lithium-heparin–containing tubes on days 1, 14, 21, 36, 49, and 56 (see text, Supplemental Digital Content 4, text describing blood sampling and analysis, available at https://links.lww.com/PR9/A195).
On days 1 and 36 (first day of treatment periods), samples were collected in the morning just before the intake of the first tablet and then subsequently at 15, 30, 45, and 60 minutes after tablet intake. On days 14, 21, 49, and 56, medication was taken in the morning, and samples were collected during the clinical visit 1 to 3 hours later.
2.11. Adverse eventsDefinitions, monitoring, and reporting procedures are described in Supplemental Digital Content 5 (see text, Supplemental Digital Content 5, text describing adverse events, available at https://links.lww.com/PR9/A195).
2.11.1. SafetyLow-dose naltrexone is considered safe to administer. Four studies19,55–57 only reported mild adverse events. The following adverse events were specifically asked for and reported: sleep disturbances, vivid dreams, nausea, diarrhea, headache, and tiredness.
2.12. Statistics 2.12.1. Statistical significanceThe authors are aware of the discussions concerning the indiscriminate use of P values as an absolute mean of null hypothesis testing.1,49,50 In this article, the term statistical significance was avoided. The advice “correct and careful interpretation of statistical tests demands examining the sizes of effect estimates and confidence limits, as well as precise P values (not just whether P values are above or below 0.05 or some other threshold)” was generally followed.6,17,20
2.12.2. Sample size estimatesThe calculation is based on FIQ data from Younger et al.,56 where mean values (SD) in the LDN group of 28.8 (12.5)% and in the placebo group of 18.0 (14.6)% are given for pain reduction, which gives an effect size (ES) of 0.61 (GPower*3.1.9.2, Kiel University, Germany).
The sample size estimates were based on a 1% chance of type Ⅰ errors (α = 0.01), 10% chance of type Ⅱ errors (β = 0.10), nonparametric distribution (ARE-correction; paired analysis with Wilcoxon signed-rank test), and an estimated correlation coefficient (r) between the treatments of 0.3. The estimated sample size per center was 51, allowing complete analyses to be performed at each center (see text, Supplemental Digital Content 6, text describing statistics, available at https://links.lww.com/PR9/A195).
2.12.3. Statistical data processingOur analyses focused on measuring the pharmacodynamics effects of LDN compared with placebo on a number of primary and secondary outcomes. We exploited our access to both baseline and outcome measures for all individuals under both active treatment and placebo. This allowed us to perform paired tests. First, baseline (BA) and outcome (OA) measures were transformed into measures of changes (Δ) for all variables (v) and for all individuals (i) under treatment with LDN or placebo:Δvi=viOA−viBA.
To assess the pharmacodynamics effects of LDN, the differences between LDN treatment and placebo were examined (see text, Supplemental Digital Content 6, text describing statistics, available at https://links.lww.com/PR9/A195). Formally testing the null hypotheses of no effects, paired Wilcoxon signed-rank tests were used to report the associated P values and appropriate ES. For completeness, the results of the corresponding parametric tests were reported in tabular form (mean [SD], P, 95% confidence interval [CI], effect sizes [ESs]). Data are reported as median (IQR) unless otherwise stated.
3. Results 3.1. PatientsA total of 151 patients with FM were assessed for eligibility, and 58 patients with FM were included and randomized. Sixty patients declined to participate mostly due to time requirement, and 33 did not meet inclusion criteria. Two patients with FM dropped out due to adverse events (nausea, vomiting) immediately after the treatment started. One patient dropped out due to concomitant acute illness, and 3 patients with FM did not state the withdrawal reason. Fifty-two patients with FM fulfilled the study per protocol. The first patient was included in May 2016, and the last patient visit was in December 2019. Inclusion was evenly distributed over time.
3.1.1. Concomitant medication and demographicsDemographic data and use of concomitant medication are described in Table 3 (see Table 3 describing demographic) and Table 4 (see Table 4 describing concomitant medication), respectively. The patients with FM continued the medication in stable doses during the study.
Table 3 - Demographics data. Demographics Value Mean Male/female, n 6/46 — Age min/max, y 24/66 50.4 BMI min/max, kg/m2 20/41 31.2 Weight min/max, kg 52/130 86.5*Number of patients with intake.
†Daily dose.
‡Prn.
§Fixed daily dose.
Adverse events (see text, Supplemental Digital Content 5, text describing adverse events, available at https://links.lww.com/PR9/A195) were registered at visit days in both treatment periods. Headache, fatigue, nausea, and dizziness were registered in both treatment periods by a small number of patients with FM, and all adverse events were classified as minor (See Table 5 describing adverse events). The 2 patients with FM who dropped out immediately after the start of the treatment experienced minor adverse events.
Table 5 - Patients reported adverse events. Adverse event LDN treatment (n) Placebo treatment (n) Headache 9 9 Fatigue 9 4 Nausea 8 5 Dizziness 7 4 Stomach ache 3 4 Diarrhea 3 2 Constipation 2 2 Blurry vision 1 2 Worsened sleep 0 3 Improved sleep 1 3 Pain more intense 2 1Adverse events reported in dairy by patient during each treatment period (n = 52). All adverse events were classified as minor, and no serious adverse events or suspected unexpected adverse reactions occurred.
LDN, low-dose naltrexone.
Baseline and outcome scores for FIQR were obtained under both LDN and placebo treatment (n = 50). The differences were −1.65 (18.55) (see Fig. 3 dot-line diagram showing scores). The Wilcoxon signed-rank test did not indicate any difference between LDN and placebo (ES = 0.15, CI = −6.72 to 2.15; P = 0.30; see Table 6 describing FIQR results). The random effects model did not indicate any difference between LDN and placebo (conditional mean difference −2.50, P = 0.34) (see text, Supplemental Digital Content 6, text describing statistical data processing, available at https://links.lww.com/PR9/A195; see Table 7 describing absolute measures).
 Figure 3.:
Figure 3.: Dot-line diagram presenting change in FIQR score (left panel) and SPIR score (right panel) after LDN treatment and placebo treatment. Each line represents a patient, mean and median values for all patients shown in bottom of figure. Negative values (below 0) indicate improvements. Black dashed line indicates mean value at LDN and placebo treatment. FIQR, Fibromyalgia Impact Questionnaire revised; LDN, low-dose naltrexone; SPIR, summed pain intensity ratings.
Table 6 - Changes in primary outcome variables from low-dose naltrexone treatment to placebo treatment. Outcome n Median IQR P CI (95%) ES Mean SD P CI (95%) Cohen d (ES) FIQR (score) 50 −1.65 18.55 0.30 −6.72, 2.15 0.15 −2.19 20.40 0.45 −7.98, 3.61 −0.11 SPIR (score) 45 −0.33 6.33 0.40 −2.17, 0.92 0.13 −0.23 6.48 0.24 −2.18, 1.72 −0.04ES, effect size; FIQR, Fibromyalgia Impact Questionnaire revised; IQR, interquartile range; n, number of patients; SPIR, summed pain intensity rating.
Comments (0)