
Remember me

We report five unrelated individuals (five females) from separate tertiary centres, each with molecularly confirmed diagnoses of APS1. The median age at first ocular symptom or sign was 14 years (range 3–20 years) and at most recent follow-up was 33 years (range, 19–69 years), with a median ophthalmic follow-up duration of 14 years (range, 5–57 years). Case 2 and 5 have previously reported data [7,8,9,10]. Three distinct AIRE variants were identified across the cohort: the recurrent frameshift variant (NM_000383.4) c.967_979del; p.(Leu323Serfs*51) the founder nonsense variant c.769C>T; p.(Arg257*), and a previously reported deletion c.1265del; p.(Pro422Leufs*58) [11, 12]. Retinal involvement was observed in four patients. Notably, one patient (Case 1) exhibited no anatomical or functional evidence of retinopathy over five years of high-resolution imaging and electrophysiological surveillance.
Key systemic and ocular features for all five patients are summarised in Table 1.
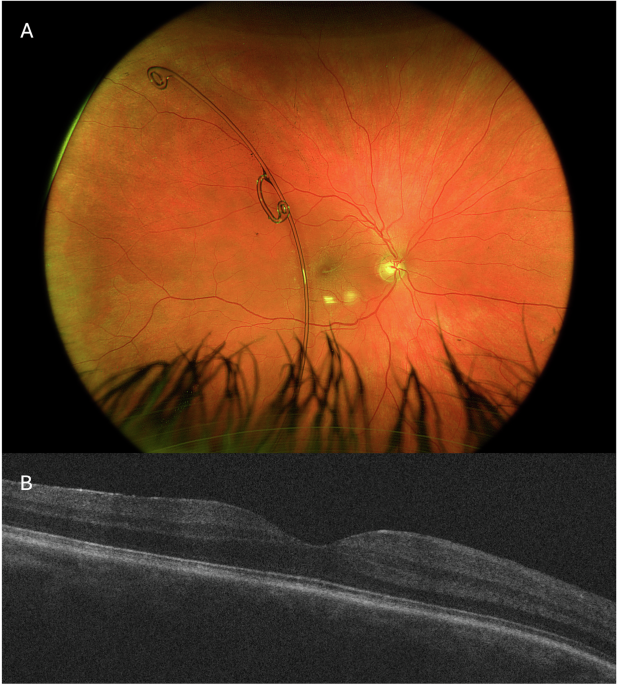
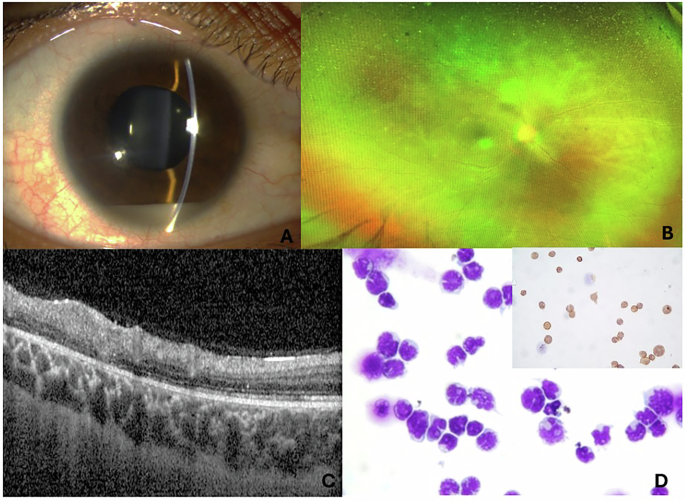
Table 1 Systemic profile, management and follow up of the five patients in our case series.Case 1This patient, a 19-year-old woman, fulfilled the diagnostic triad of APS1 in childhood. Genetic testing confirmed a biallelic homozygous pathogenic AIRE frameshift deletion variant: c.967_979del; p.(Leu323Serfs*51), inherited in trans from both parents, consistent with autosomal recessive autoimmune polyendocrinopathy with candidiasis and ectodermal dysplasia. At 14 years old, she was diagnosed with idiopathic intracranial hypertension (IIH). Fundoscopy revealed bilateral optic disc hyperaemia, but foveal reflexes were preserved, and best-corrected visual acuity (BCVA) was 20/20 in both eyes. Other than disc swelling, ocular examination and multimodal retinal imaging remained normal (Supplementary Fig. 1). Peripapillary retinal nerve fibre layer (RNFL) OCT revealed increased thickness (mean: 164 µm OD, 141 µm OS).
Her IIH was medically managed with acetazolamide under neurology supervision. Serial OCT-RNFL scans documented progressive normalisation of RNFL thickness, followed by mild thinning to the low-normal range (final global RNFL: 103 µm OD, 99 µm OS). No visual field defects were detected. Throughout five years of surveillance, BCVA has remained 20/20 bilaterally. She reports no nyctalopia, photopsia, or colour vision disturbances. Full-field electroretinogram (ERG) at the age of 18 years was entirely within reference limits.
Case 2This was a 25-year-old woman with biallelic disease-causing AIRE variants, specifically the founder nonsense variant c.769C>T; p.(Arg257*). She had been diagnosed clinically at 11 years old with the classical APS1 triad. The abnormal areas of histology were previously reported [8]. Long-term corticosteroid replacement with hydrocortisone and fludrocortisone was initiated at diagnosis; no additional immunosuppressive therapy was employed.
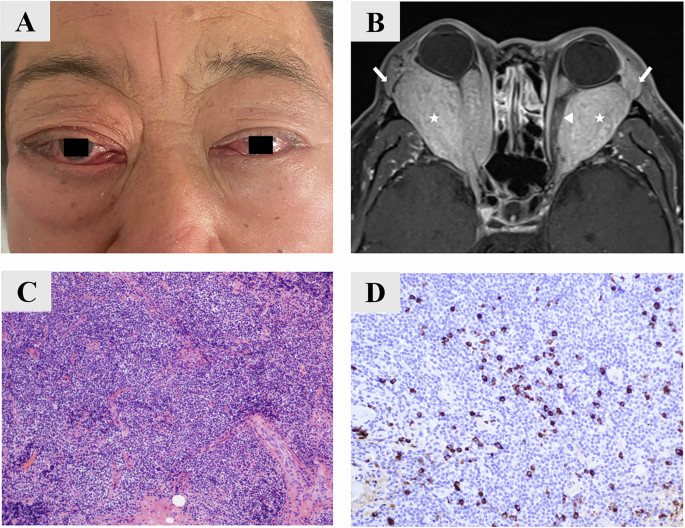
At 23 years old, the patient reported blurred vision for some years, and ocular examination revealed corneal epithelial erosions and cataracts. Visual acuities were 20/125 (right) and 20/50 (left). Fundus examination revealed right optic disc swelling (MRI showed signs of raised intracranial pressure), subtle peripapillary retinal pigment epithelium (RPE) mottling and peripheral pigment clumping (fundus photographs from the age of 23 years are in the previous report [8]), with preserved macular appearance. She was treated with azetazolamide and underwent right eye cataract surgery and reported improvement in vision. Her most recent recorded visual acuities were 20/40 in both eyes. The patient was subsequently lost to follow up and died at 25 years of age from cardiac arrest due to electrolyte abnormalities.
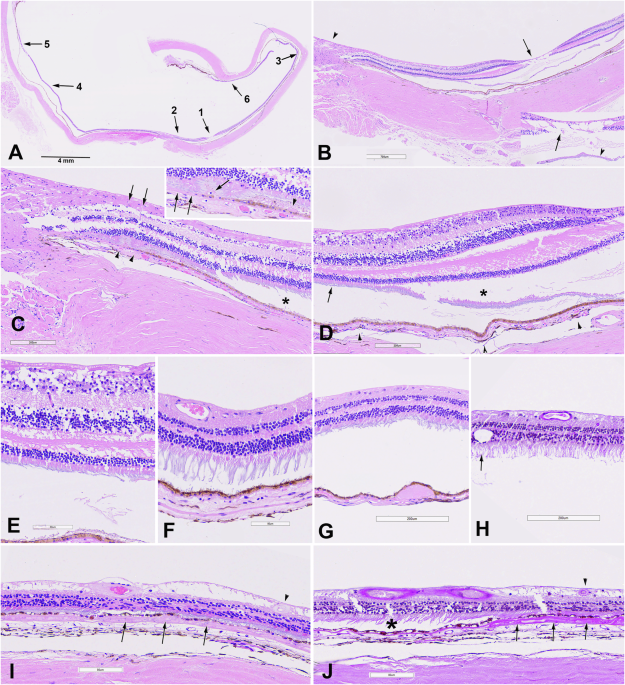
As previously reported, gross examination of both globes revealed a pattern of zones of outer retinal atrophy, some of which were contiguous with the optic disc, sparing the fovea and histological analysis of the affected areas demonstrated complete loss of photoreceptors, RPE, and choriocapillaris, with RPE hyperplasia, and reactive gliosis reminiscent of retinitis pigmentosa [8]. We now report and demonstrate that the adjacent extra-lesional retina was structurally preserved in the left eye (Fig. 1, stained with haematoxylin and eosin). Importantly, there was no evidence of active intraocular inflammation - no lymphocytic infiltration or granuloma formation - suggesting the absence of high-grade uveitis at the time of death. Furthermore, there was a type 1 choroidal neovascular membrane in the peripheral temporal retina in close proximity to the area with photoreceptor loss (Fig. 1J).
Fig. 1: Case 2, histopathologic findings of the left retina, RPE, and choroid outside of pseudo-retinitis pigmentosa changes reported previously [18].![Fig. 1: Case 2, histopathologic findings of the left retina, RPE, and choroid outside of pseudo-retinitis pigmentosa changes reported previously [18].](https://www.nature.com//media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs41433-026-04365-9/MediaObjects/41433_2026_4365_Fig1_HTML.png)
A Overview of posterior segment at scanning magnification with regions of interest magnified in panels (B–J). B Higher magnification of region designated as “1” and “2” in (A) through fovea (arrow) and optic nerve (arrowhead). Inset: Higher magnification of fovea with likely artefactual pseudo-cystoid changes in the retina (arrow) and artefactual detachment of photoreceptors (arrowhead). C Higher magnification of optic nerve and adjacent temporal retina shows mild retinal nerve fibre layer (RNFL) thinning associated with mild gliosis (arrows); RPE is focally atrophic and hypopigmented (arrowheads) and overlying retina shows outer retinal folds/tubulations. The outer retina is otherwise well-preserved and is focally artifactually detached (asterisk). The choroid shows fewer melanocytes, compared to choroid in other locations. The RPE and choroid changes correspond to the atrophic pigmentary changes described clinically [8]. The inset highlights region of RPE atrophy and hypopigmentation (2 arrows), retinal tubulations (single arrow), and subretinal proteinaceous debris (arrowhead), compatible with history of optic disc oedema [8]. D Higher magnification of the nasal fovea shows largely unremarkable retina with mild RNFL thinning, well-preserved outer retina including photoreceptor segments (arrow), artefactual retinal detachment, and artefactual detachment of photoreceptor segments (asterisk). Note more pronounced pigmentation in the choroid (arrowheads). E Higher magnification of region designated with arrow in (D). F Post-equatorial temporal retina, corresponding to region “3” in (A) is largely unremarkable with preserved photoreceptor segments and other layers. G Nasal equatorial posterior segment, corresponding to region “4” in (A) the figure (A) shows a soft drusenoid sub-RPE deposit. Subtle degenerative changes in outer photoreceptor segments in overlying artefactually detached retina may also be an artefact. H Peripheral nasal retina corresponding to region “5” in (A) close to ora serrata, shows the posterior edge of typical peripheral cystoid degeneration (arrow) and is otherwise unremarkable with good preservation of photoreceptor segments and artefactual detachment. I Peripheral temporal retina, corresponding to region “6” in (A) shows focal chorioretinal adhesion with patchy outer retinal and RPE atrophy, choriocapillaris atrophy, and neovascularisation within diffuse soft drusen (arrows). Sclerosed vessel is present in the inner retina (arrowhead) and RNFL shows degenerative changes. J Corresponding Periodic acid-Schiff-stained preparation shows neovascular membrane within diffuse drusen (arrows) with overlying patchy RPE and outer retinal atrophy, sclerosed retinal vessel (arrowhead) and underlying choriocapillaris atrophy. The more posterior unremarkable retina is artefactually detached (asterisk). [haematoxylin-eosin (A–G, I) PAS (H, J)].
This case illustrates a manifestation of AIRE-associated retinopathy characterised by localised, progressive zonal degeneration with preserved left eye central vision until death, yet histologically severe photoreceptor and RPE loss in affected areas.
Case 3This case involves a 60-year-old woman with early-onset nyctalopia, first noted at 3 years old, followed by bilateral cataract needling at the age of 4 years due to hypocalcaemia-induced cataracts, resulting in aphakia. By 6 years of age, she had developed multiple systemic features consistent with APS1, including congenital hypoparathyroidism, Addison disease (managed with hydrocortisone and fludrocortisone), type 1 diabetes mellitus, nephrolithiasis-associated renal impairment, and chronic mucocutaneous candidiasis. She was not receiving systemic immunosuppression.
Molecular genetic testing revealed two pathogenic AIRE frameshifting deletions: c.967_979del; p.(Leu323Serfs*51) and c.1265del; p.(Pro422Leufs*58). Although phasing could not be established, the clinical phenotype was fully consistent with autosomal recessive APS1.
The patient remained ophthalmologically stable until 25 years old, when anterior and intermediate uveitis were diagnosed, treated with periocular corticosteroid therapy. By the age of 31 years, she had been diagnosed with “retinitis pigmentosa” at an external clinic; however, her phenotype evolved to asymmetric multifocal chorioretinal scarring with peripheral pigment clumping. Visual acuity with contact lenses was 20/30 bilaterally by 40-years-old.
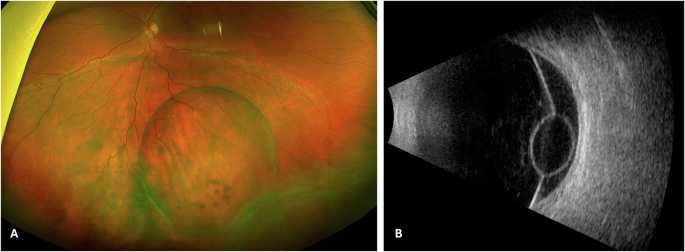
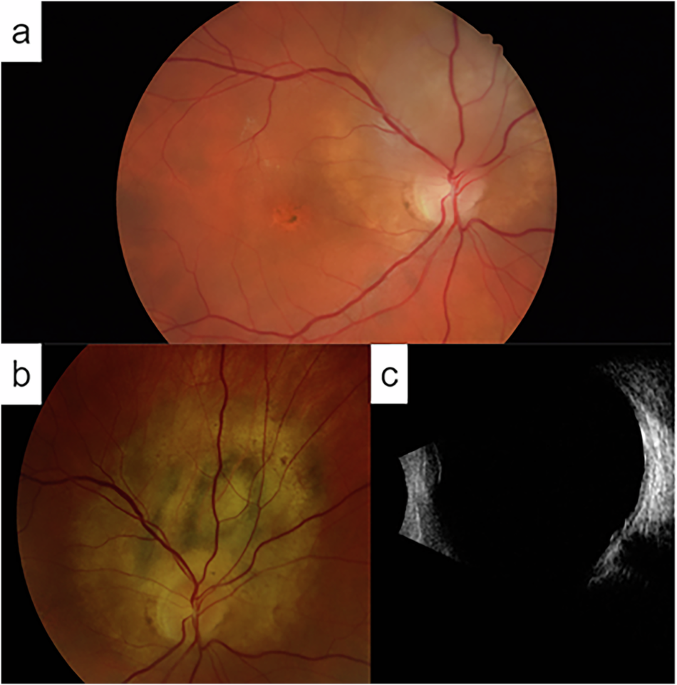
Retinal imaging, at 52 and 60 years of age, is shown in Fig. 2A–D, aged 52; E, F, aged 60. At 52 years old, multifocal areas of chorioretinal scarring were visible clinically and on FAF (asymmetric and more confluent in the left eye), with fluorescein angiography showing areas of nonperfusion and disc leakage. Macular OCT showed temporal parafoveal outer retinal atrophy in the right eye and an epiretinal membrane in the left. She subsequently developed bilateral granulomatous uveitis. Ocular hypertension was diagnosed at 54 years old.
Fig. 2: Multimodal imaging from Case 3.
Ultra-widefield pseudocolour (A) autofluorescence (B) fluorescein angiography (C) and OCT images (D) from right and left eyes aged 52. The fluorescein angiograms were at 31 s and 2 min 10 s for right and left eyes, respectively. Follow-up ultra-widefield pseudocolour (E) and OCT (F) images aged 60.
Surgical interventions included right pars plana vitrectomy at 55 years old for vitreous debris and left vitrectomy with epiretinal membrane peel at 59 years old. At most recent follow up (60 years old), she maintains visual acuity of 20/30 (OD) and 20/40 (OS) with contact lens correction (imaging shown in lower panels of Fig. 2).
Systemic immunosuppression was not used for this individual, management included topical lubricants (Hylo-Forte) and intraocular pressure control with topical bimatoprost/timolol eye drops. Vitritis flares were treated with sub-Tenon’s triamcinolone (5 injections between 46 and 53 years of age). Intraoperative PCR testing for viral pathogens during vitrectomy was negative. Anti-retinal antibody testing was negative (recoverin, carbonic anhydrase II and alpha enolase).
Case 4This case is a 69-year-old woman who presented with nyctalopia at 12 years old. She was subsequently followed under the diagnosis of “retinitis pigmentosa/retinal dystrophy”. Systemic manifestations included Addison disease (on hydrocortisone and fludrocortisone since 12 years old), chronic mucocutaneous candidiasis, hypoparathyroidism with intermittent hypoglycaemia, primary ovarian failure, and obesity, consistent with APS1.
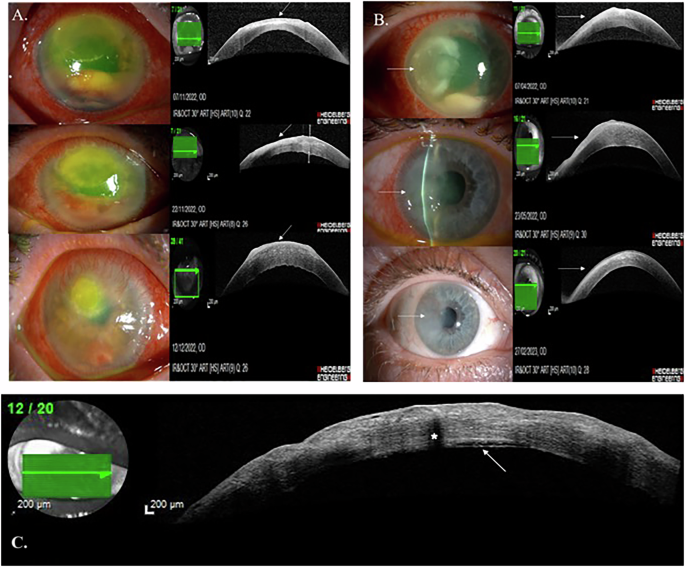
At 47 years old, her best-corrected visual acuity (BCVA) was 6/12 (20/40) in both eyes. Fundoscopy revealed paravascular pigment clumping and chorioretinal atrophy radiating from the optic disc. Her pedigree and fundus imaging (from 52 to 68 years of age) are shown in Fig. 3.
Fig. 3: Pedigree & multimodal imaging from Case 4.
A Family pedigree. B Left eye colour fundus image aged 52. Colour fundus (C) autofluorescence (D) and OCT (E, F) images from right and left eyes aged 61. Colour fundus (G) and autofluorescence (H, I) images from right and left eyes aged 63. Colour fundus (J) autofluorescence (K) and OCT (L, M) images from right and left eyes aged 68.
By 68 years old, she retained only light perception in both eyes (bilaterally pseudophakic). She was on systemic corticosteroids for adrenal insufficiency, and no additional immunosuppressive treatment was initiated.
The patient previously underwent targeted AIRE gene testing, which identified a homozygous pathogenic AIRE frameshift deletion: c.967_979del; p.(Leu323Serfs*5), a known disease-causing variant [13]. She also underwent whole genome sequencing, and no additional pathogenic genotypes were identified in known IRD genes.
Case 5This case describes a 33-year-old woman with loss-of-function AIRE variants (Table 1). Her systemic phenotype conforms to the classical triad of APS1 and includes autoimmune ovarian failure and ectodermal features.
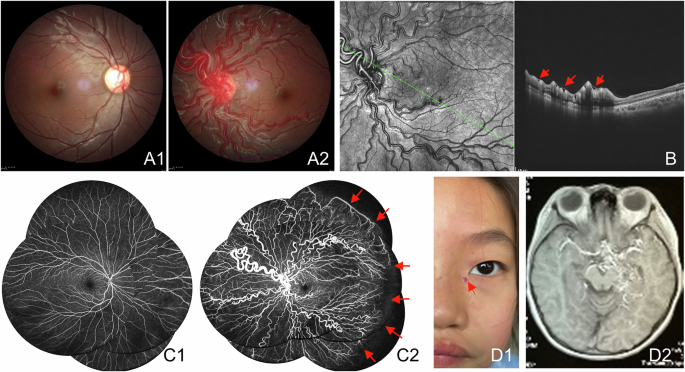
At 19 years old, she developed concentric visual field constriction. Fundoscopic examination revealed dense mid-peripheral bone-spicule pigmentation with relative preservation of the macula. OCT demonstrated parafoveal loss of the ellipsoid and external limiting membranes, with an intact foveal centre (Fig. 4C). FAF showed a hyperautofluorescent ring - resembling the “Robson ring” - demarcating the outer boundary of intact photoreceptors (Fig. 4A, B), seen in rod-cone dystrophies [14]. Progression of retinal degeneration was observed over time, with no evidence of anterior segment inflammation or keratopathy.
Fig. 4: Multimodal imaging from Case 5.
A Ultra-widefield pseudocolour imaging from both eyes. B Fundus autofluorescence showing characteristic hyperautofluorescent ring (“Robson ring”). C OCT demonstrating outer retinal thinning with preserved fovea from both eyes.
This case has previously been reported by Breunig et al. in 2013. However, from 2019 to 2023, the patient received B-cell-depleting therapy with rituximab, administered biannually [7]. Visual acuity remained stable under this regimen. However, rituximab was discontinued due to episodes of neutropenia. Subsequent treatment with adalimumab was curtailed following the emergence of psoriatic dermatoses.
The patient is currently maintained on cyclosporine (100 mg daily), mycophenolate mofetil (750 mg twice daily), and low-dose prednisone. Best-corrected visual acuities are 20/25 (OD) and 20/30 (OS). Despite treatment, OCT reveals ongoing parafoveal thinning.
Previously reported casesA literature search revealed 9 previous publications (reporting a total of 34 cases) describing retinopathy in association with APS1. Demographics, phenotypic features, genotypes and clinical course are summarised in Supplementary Table 1.
Comments (0)