
Remember me
Objective:
To investigate the associations of C-reactive protein (CRP), triglyceride–glucose (TyG) index, and their composite—the CRP–TyG index (CTI)—with sequential trajectories within the cardiovascular–renal–diabetes (CRD) cluster, including incident coronary artery disease (CAD), type 2 diabetes mellitus (T2DM), chronic kidney disease (CKD), and multimorbidity.
Methods:
We analyzed 333,698 participants from the UK Biobank who were free of CAD, T2DM, and CKD at baseline. CRP and TyG were assessed individually and jointly through the CTI. Multistate Cox models were applied to evaluate six predefined transitions within the CRD cluster, with multimorbidity defined as the coexistence of at least two of CAD, T2DM, and CKD. Potential nonlinearity was assessed using restricted cubic splines, and time-dependent effects were examined with piecewise analyses. Joint exposure analyses assessed synergistic effects of CRP and TyG, while receiver operating characteristic (ROC) curves compared the predictive performance of CRP, TyG, their combination, and CTI. Subgroup and sensitivity analyses were performed to test heterogeneity and robustness.
Results:
During a median follow-up of 15.31 years (IQR, 14.54–16.03 years), CTI was consistently associated with higher risks of CAD (HR per 1-SD: 1.23, 95% CI: 1.21–1.25), T2DM (1.88, 95% CI: 1.84–1.92), CKD (1.22, 95% CI: 1.19–1.25), and multimorbidity (1.59, 95% CI: 1.55–1.64), outperforming CRP and TyG individually. CTI exhibited trajectory-specific heterogeneity, with nonlinear associations observed in most baseline-to-disease transitions (P for nonlinearity <0.05) and predominantly linear associations during progression from single diseases to multimorbidity (all P for nonlinearity >0.05). Moreover, higher CTI was associated with a time-dependent cumulative increase in multimorbidity risk. In joint exposure analyses, participants with both high CRP and high TyG had the greatest risks across outcomes, including CAD (HR 1.48, 95% CI: 1.40–1.56), CKD (HR 1.52, 95% CI: 1.40–1.64), T2DM (HR 3.63, 95% CI: 3.35–3.93), and multimorbidity (HR 2.64, 95% CI: 2.47–2.82).
Conclusions:
CRP, TyG, and CTI were strongly associated with both the onset and progression of the cardiovascular–renal–diabetes cluster. By integrating metabolic and inflammatory risk signals, CTI outperformed its individual components, underscoring its clinical utility for refined risk stratification and for guiding early, stage-specific prevention strategies.

CTI integrates metabolic (TyG) and inflammatory (CRP) signals to capture shared mechanisms within the cardiovascular–renal–diabetes cluster. Multistate analyses depict transitions from baseline to first incident CAD, T2DM, or CKD and subsequent progression to multimorbidity, showing higher CTI-associated risks across trajectories and improved discrimination compared with CRP, TyG, and their combination. CRP, C-reactive protein; TyG, triglyceride–glucose index; CTI, C-reactive protein–triglyceride–glucose index; T2DM, type 2 diabetes mellitus; CAD, coronary artery disease; CKD, chronic kidney disease.
HighlightsCTI outperformed CRP, TyG, and their combination in predicting CAD, T2DM, CKD, and multimorbidity.
CTI-related risks intensified over time, with multimorbidity becoming markedly elevated beyond 5 years.
CTI provides refined risk stratification and supports stage-specific prevention in the CRD cluster.
IntroductionCoronary artery disease (CAD), type 2 diabetes mellitus (T2DM), and chronic kidney disease (CKD) are among the most prevalent metabolic disorders worldwide. Ischemic heart disease, the dominant clinical manifestation of coronary artery disease, remains the leading single cause of death worldwide, accounting for approximately 9.1 million deaths in 2021, as reported by the World Health Organization (1). Global Burden of Disease (GBD) estimates further suggest that about 197 million people were living with ischemic heart disease in 2019 (2). Diabetes affected an estimated 589 million adults aged 20–79 years worldwide and is projected to increase substantially; it was associated with approximately 3.4 million deaths in 2024, according to the International Diabetes Federation (3). Chronic kidney disease also represents a major global health burden, affecting more than 800 million people globally, often reported as approximately 850 million in global kidney health reports, and accounting for an estimated 1.48 million deaths in 2023 based on GBD-aligned estimates (4). With population aging and the rising burden of metabolic diseases, their coexistence has emerged as a major public health challenge (5, 6). Epidemiologic data demonstrate substantial overlap: the presence of one condition markedly increases the risk of the others and worsens outcomes. Shared pathophysiologic pathways—metabolic dysfunction and chronic inflammation—interact bidirectionally, reinforcing a vicious cycle of excess morbidity and mortality (6). Although distinct entities, CAD, CKD, and T2DM converge on these mechanisms and mutually accelerate disease progression.
Insulin resistance (IR), the hallmark of T2DM, represents a key metabolic link. IR promotes atherogenesis through dyslipidemia, increasing CAD risk, and activates the renin–angiotensin–aldosterone system, predisposing to CKD. Conversely, myocardial ischemia in CAD exacerbates systemic metabolic dysregulation and IR, while CKD-related uremic toxins impair insulin signaling, further worsening IR (7–9). Chronic inflammation similarly drives cross-disease progression. In T2DM, adipose-derived cytokines (e.g., TNF-α, IL-6) impair β-cell function, aggravate glycemic dysregulation, and promote plaque instability, elevating CAD risk. In CAD, plaque rupture provokes systemic inflammation that accelerates renal fibrosis and CKD progression. In turn, CKD-associated systemic inflammation intensifies coronary plaque inflammation, hastening CAD (9–12). These intersecting mechanisms underpin the marked tendency toward comorbidity. Mapping the dynamic trajectories of the cardiovascular–renal–diabetes (CRD) cluster—from disease-free status to single conditions to multimorbidity—and identifying biomarkers driving these transitions are essential for early prevention and intervention.
The triglyceride–glucose (TyG) index is a well established surrogate for IR, valued for its simplicity and reliance on routine laboratory measures, enabling broad use in large-scale studies (13–15). C-reactive protein (CRP), a sensitive marker of low-grade inflammation, has likewise been associated with CAD, CKD, and T2DM. However, each marker alone has limitations; TyG captures metabolic dysfunction but not inflammation, whereas CRP reflects inflammation but not metabolic abnormalities (13, 16, 17). Recognizing the interplay between IR and inflammation, the CRP–TyG index (CTI) was proposed as a composite marker integrating both pathways. Preliminary evidence indicates that CTI offers superior predictive performance for CAD, CKD, and T2DM compared with TyG or CRP alone (18–21).
Nevertheless, important gaps remain. First, CTI has not been systematically evaluated across disease progression, especially from single disease to multimorbidity. Second, potential heterogeneity in progression trajectories by initial disease is poorly understood. Third, reliance on conventional Cox regression models, which precludes assessment of sequential disease ordering and competing risks, potentially leading to biased risk estimates.
To address these gaps, we applied multistate models in a large prospective cohort to evaluate associations of TyG, CRP, and CTI with transitions within the CRD cluster, aiming to clarify their roles in disease dynamics and to inform risk stratification and stage-specific prevention strategies.
MethodStudy populationThis analysis was conducted within the UK Biobank, a large prospective cohort that recruited 502,128 participants aged 40–69 years between 2006 and 2010. At baseline, sociodemographic characteristics, lifestyle factors, physical measurements, and biological samples were collected using standardized protocols. Health outcomes were identified through linkage to national death registries and hospital episode statistics, with detailed study design reported elsewhere (22).
Participants with prevalent CAD (n=27,620), heart failure (n=828), valvular heart disease (n=2,534), cardiomyopathy (n=381), arrhythmias (n=10,642), stroke (n=5,801), T2DM (n=19,580), or CKD (n=3,535) were excluded. The diagnostic details are provided in Supplementary Table 1. We also excluded individuals with pregnancy (n=140), cancer (n=40,301), and those lacking complete biomarker data required for CTI calculation (n=57,068).The final analytic sample comprised 333,698 participants free of cardiovascular disease, diabetes, chronic kidney disease, cancer, or pregnancy at baseline.
The UK Biobank received ethical approval from the North West Multi-Centre Research Ethics Committee (reference 21/NW/0157), and all participants provided written informed consent. This study adhered to the STROBE guidelines for reporting observational research.
Calculation of the TyG index and CTICRP was quantified using an immunoturbidimetric assay on the Beckman Coulter AU5800 platform (Beckman Coulter, UK). Triglycerides and plasma glucose were measured using enzymatic methods on the same analyzer.
The CTI was developed to jointly capture systemic inflammation and insulin resistance, integrating CRP and the TyG index (18, 23). The TyG index was calculated as: TyG=ln(TG [mg/dL] × plasma glucose [mg/dL])/2, where plasma glucose refers to random plasma glucose in the UK Biobank. The CTI was then derived according to the established formula: CTI = 0.412×ln(CRP [mg/L])+TyG.
Ascertainment of CAD, T2DM, CKD, and mortalityIncident CAD, T2DM, and CKD were identified using the UK Biobank First Occurrences dataset (Category 1712), which integrates data from primary care records, hospital admissions, death registries, and self-reports, mapped to 3-character International Classification of Diseases, Tenth Revision (ICD-10) codes. CAD was defined by ICD-10 codes I20–I25, T2DM by E11, and CKD by N18.
All-cause mortality was determined through linkage to national death registries, including NHS Digital (England and Wales) and the NHS Central Register (Scotland), with dates of death obtained from official death certificates.
Outcome data were available through August 1, 2024, for CAD and T2DM; July 1, 2024, for CKD; and July 8, 2024, for mortality. To ensure consistency across outcomes, July 1, 2024, was chosen as the common study end date. Participants were followed from the date of enrollment until the occurrence of multimorbidity, death, loss to follow-up, or July 1, 2024, whichever occurred first.
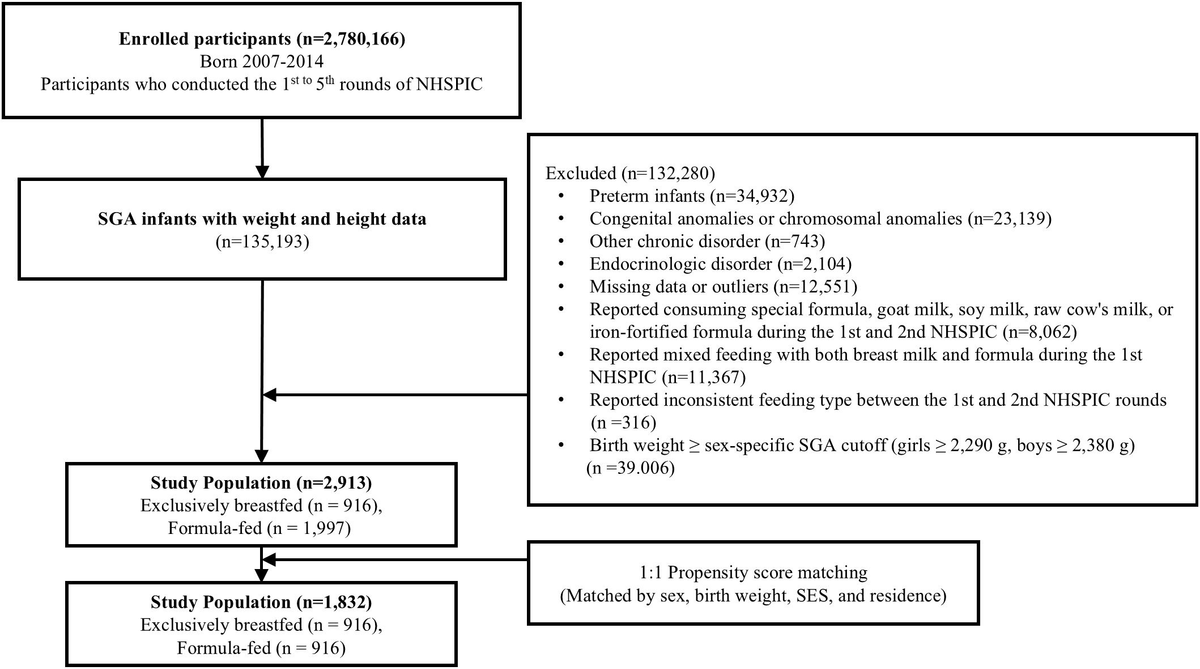
Primary outcome and transition pathwaysThe primary outcome was the development of multimorbidity, defined as the occurrence of at least two of the following conditions: CAD, T2DM, and CKD. Disease progression was evaluated through six transition pathways: (1) baseline to incident CAD, (2) baseline to incident T2DM, (3) baseline to incident CKD, (4) CAD to multimorbidity, (5) T2DM to multimorbidity, and (6) CKD to multimorbidity (Figure 1).

Transition pathways in the cardiovascular–renal–diabetes (CRD) cluster. Six predefined transitions were modeled: baseline to CAD (transition 1), baseline to T2DM (transition 2), baseline to CKD (transition 3), CAD to multimorbidity (transition 4), T2DM to multimorbidity (transition 5), and CKD to multimorbidity (transition 6). Multimorbidity was defined as the coexistence of at least two conditions among CAD, T2DM, and CKD.
Assessment of covariatesBaseline characteristics were obtained from standardized touchscreen questionnaires, nurse-led interviews, physical measurements, and biochemical assays. Demographic factors included age, sex, and self-reported race (White vs. non-White). Socioeconomic status was assessed using the Townsend Deprivation Index (TDI, a validated area-based measure of material deprivation), education level (college degree or equivalent vs. high school or below), and average household income (<£30,999 or ≥£30,999). Lifestyle factors comprised physical activity (classified as low, moderate, or high according to the International Physical Activity Questionnaire), smoking status (active or non-active), alcohol consumption (active or non-active). Furthermore, a cumulative dietary risk score was constructed from the baseline touch-screen questionnaire to characterize individual-level dietary patterns. This composite score summarized an overall adverse dietary pattern, capturing higher intake of high-fat and saturated-fat–rich foods such as processed and red meat, full-cream milk, and spreads, as well as lower intake of protective foods such as fruits, vegetables, and fish. It also incorporated other diet-related behaviors, including cereal, salt, and water intake. Each adverse dietary component contributed one point to the cumulative score, resulting in a scale from 0 (most favorable) to 9 (least favorable). The score was classified as low (≤6 points) or high (≥7 points), with details provided in Supplementary Table 2 (24). Clinical variables included body mass index (BMI), fasting duration (hours since last meal), glycated hemoglobin (HbA1c), hypertension, and use of lipid-lowering or antihypertensive agents. Biochemical analytes were quantified with standardized enzymatic assays on the Beckman Coulter AU5800 platform. For all categorical variables, responses recorded as “prefer not to answer,” “do not know,” or “missing” were classified as a separate category.
To identify potential confounders of the associations between CTI and outcomes, we constructed a directed acyclic graph (DAG) using Dagitty (www.dagitty.net), in line with contemporary causal inference practices (25). The DAG identified a minimally sufficient adjustment set comprising age, sex, BMI, race, physical activity, hypertension, cumulative dietary risk score, smoking status, alcohol consumption, lipid-lowering therapy, HbA1c, and fasting hours (Supplementary Figure 1). Cholesterol levels and antihypertensive therapy were regarded as potential mediators rather than confounders: cholesterol reflects downstream metabolic alterations along the pathway from CTI-related dysmetabolism to clinical outcomes, whereas antihypertensive therapy represents a treatment response to elevated blood pressure. To avoid overadjustment bias and preserve estimates of the total effect of CTI, these variables were excluded from the final models.
In addition, covariates were evaluated empirically and retained if they satisfied at least one of two criteria for any outcome: (1) inducing >10% change in the β coefficient for the CTI–outcome association, or (2) demonstrating an independent association with the outcome at P < 0.1 (26, 27). To ensure consistency and minimize residual confounding, all covariates meeting either criterion were included in the final models (Supplementary Table 3).
Statistical analysisMissing data for continuous variables were imputed using mean values, and categorical variables with missing responses (e.g., “prefer not to answer”) were grouped into a separate category. Continuous variables are reported as means with standard deviations (SDs), and categorical variables as counts and percentages. Exposures of interest included CRP, the TyG index, and CTI. To ensure comparability across measurement scales, all exposures were standardized as Z-scores (mean = 0, SD = 1). Associations of CRP, TyG, and CTI with incident CAD, T2DM, CKD, and multimorbidity were initially evaluated using conventional Cox proportional hazards models, with hazard ratios (HRs) and 95% confidence intervals (CIs) estimated per 1-SD increase in standardized exposures. Disease trajectories were analyzed using multistate Cox models with a clock-forward approach (mstate package), which extend conventional Cox regression by modeling sequential transitions and competing risks. Six pathways were prespecified: (1) baseline to incident CAD, (2) baseline to incident T2DM, (3) baseline to CKD, (4) CAD to multimorbidity, (5) T2DM to multimorbidity, and (6) CKD to multimorbidity. For participants who developed multiple disease states on the same date, temporal ordering could not be determined; therefore, they were excluded from the multistate models. However, these individuals remained included in the conventional Cox regression analyses.
Time-varying effects for transitions from CAD, T2DM, or CKD to multimorbidity were assessed using piecewise Cox models. Interaction terms between exposures (per 1-SD increment) and predefined follow-up intervals (0–<1, 1–<3, 3–<5, and ≥5 years) were included. Follow-up time was split with the survSplit function, and interval-specific HRs with 95% CIs were estimated and displayed in forest plots.
Because CTI was derived from CRP and TyG, receiver operating characteristic (ROC) analyses compared the predictive performance of CTI, CRP, TyG, and the combination of CRP and TyG for CRD cluster. Discrimination was quantified using the area under the curve (AUC), indicating each marker’s ability to distinguish between individuals with and without the outcome.
The TyG index was dichotomized at the cohort median (8.65) into low (<8.65) and high (≥8.65) categories. CRP was dichotomized at 5 mg/L to define chronic inflammation status, with low (<5 mg/L) and high (≥5 mg/L) groups, both coded as binary variables. Joint exposure was defined by cross-classifying these categories, yielding four groups with low CRP/low TyG as the reference. Relative risks for multimorbidity were compared across groups. Additive interaction was assessed using the relative excess risk due to interaction (RERI), attributable proportion (AP), and synergy index (S), with 95% CIs estimated via the delta method. Multiplicative interaction was tested using product terms and likelihood ratio tests. Synergy was considered present if either additive or multiplicative interaction was significant.
To investigate potential non-linear dose–response relationships between CTI and transitions within the CRD cluster, multi-state Cox models were applied for six predefined pathways. Non-linearity was assessed using restricted cubic splines (RCS), with knot numbers (3–6) selected by minimizing the Akaike Information Criterion (AIC). For each pathway, likelihood ratio tests compared spline and linear models to evaluate evidence of non-linearity. To mitigate outlier influence, CTI values were winsorized at the 1st and 99th percentiles within transition-specific risk sets. This transition-specific approach robustly captured potential non-linear effects of CTI on CRD cluster onset and progression across pathways.
Effect modification was examined by age (≤55 vs. >55 years), sex, BMI (<30 vs. ≥30 kg/m²), dietary risk score (low vs. high), hypertension (yes vs. no), and physical activity (low, moderate, high). Interactions were tested using likelihood ratio comparisons, and stratum-specific HRs with 95% CIs were reported per 1-SD increment.
Sensitivity analyses included: (1) excluding incident CAD, T2DM, or CKD cases occurring within two years of enrollment to minimize reverse causation; (2) multiple imputation of missing covariates with results pooled by Rubin’s rules; (3) restricting exposures to the 2.5th–97.5th percentiles; (4) complete-case analysis excluding participants with missing baseline data; (5) additional adjustment for low-density lipoprotein cholesterol and antihypertensive therapy; and (6) inclusion of a direct baseline-to-multimorbidity transition in the multistate framework.
All analyses were conducted in R version 4.2.3. Two-sided P values <0.05 were considered statistically significant.
ResultsA total of 333,698 participants were included, of whom 1,306 developed CAD, T2DM, or CKD on the same day, making temporal ordering indeterminate. Sample sizes for the six transition pathways were baseline to T2DM (n=13,063), baseline to CAD (n=21,677), baseline to CKD (n=9,077), T2DM to multimorbidity (n=1,685), CAD to multimorbidity (n=2,265), and CKD to multimorbidity (n=1,094). Compared with the overall cohort (mean age 55.6 years; 44.9% men), participants across all transition pathways were older, with a higher proportion of men. They exhibited adverse cardiometabolic profiles, including higher body mass index, HbA1c, CRP, CTI, and TyG levels, alongside lower HDL cholesterol. Hypertension was more common, with greater use of antihypertensive and lipid-lowering therapies.
Sociodemographic and lifestyle differences were also evident. Participants in transition pathways had higher Townsend deprivation index scores, lower household income, and lower educational attainment. They more frequently reported high dietary risk, lower physical activity, and active smoking, while non-drinking status was more common than in the overall cohort. Detailed baseline characteristics for the total population and transition-specific subgroups are shown in Table 1.
VariableTotalBaseline toBaseline characteristics of the 333,698 individuals grouped by six transition stages.
TDI, Townsend Deprivation Index; HDL, High-density lipoprotein cholesterol; LDL, Low-density lipoprotein cholesterol; TC, Total cholesterol; HbA1c, Glycated hemoglobin; CRP, C-reactive protein; CTI, C-reactive protein-triglyceride-glucose index; TyG,Triglyceride-glucose index.
Six predefined transitions were modeled: baseline to CAD (transition 1), baseline to T2DM (transition 2), baseline to CKD (transition 3), CAD to multimorbidity (transition 4), T2DM to multimorbidity (transition 5), and CKD to multimorbidity (transition 6).
Conventional cox and multistate modelsIn conventional Cox proportional hazards models, each 1-SD increment in CRP, TyG, and CTI was associated with significantly higher risks of T2DM, CAD, CKD, and multimorbidity (all P < 0.001; Table 2), exhibiting distinct risk gradients. For T2DM, the HRs (95% CI) were 1.09 (1.08–1.11) for CRP, 1.67 (1.64–1.69) for TyG, and 1.86 (1.82–1.89) for CTI (strongest association). For CAD, the corresponding HRs were 1.07 (1.06–1.08) for CRP, 1.14 (1.13–1.16) for TyG, and 1.23 (1.21–1.25) for CTI. For CKD, the HRs were similar for CRP (1.10 [1.08–1.11]) and TyG (1.10 [1.08–1.12]), but stronger for CTI (1.23 [1.21–1.26]). For multimorbidity, the HRs were 1.12 (1.11–1.14) for CRP, 1.39 (1.36–1.43) for TyG, and 1.59 (1.55–1.64) for CTI.
CaseProportion (%)HR (95% CI)P
Comments (0)