
Remember me
Introduction:
Antibiotic resistance poses a critical and escalating global health crisis, leading to higher morbidity and mortality associated with infectious diseases. This problem is significantly exacerbated by intracellular bacterial pathogens, which are often shielded from conventional antibiotics and foster the emergence of persister populations. Recently, host-directed therapy (HDT) has been emerging as a promising strategy that aims to modulate host cellular processes or immune responses to enhance bacterial clearance. Nonetheless, the inherent complexity of host biology makes identifying appropriate and safe modulators challenging, unpredictable, and highly complicated.
Methods:
Here, we present a cell-based high-throughput screen (HTS), coupled with an intracellular-induced reporter that was used to screen a library of nearly 37,000 small molecules with potentially pharmacological activity for compounds that inhibit host cell infection by intracellular pathogens.
Results and discussion:
This multistage, screening protocol resulted in the identification of eight non-cytotoxic compounds that efficiently inhibited the intracellular growth of the Gram-negative bacterium Salmonella Typhimurium in human epithelial cells by ~2.5- to 6-fold, without inhibiting Salmonella growth in culture. Five of these eight molecules were also effective in controlling the intracellular replication of Salmonella in primary mouse macrophages by 1.5- to 38-fold. Strikingly, seven hits also inhibited the intracellular growth of the Gram-positive bacterial pathogen Listeria monocytogenes in epithelial cells by 1.5- to 10-fold. The structure–activity relationship approach successfully identified chemical analogs of one hit with enhanced biological activity as infection inhibitors. Overall, we describe a robust HTS platform that can be adapted for screening of compound libraries against other pathogens, and suggest that the identified compounds are potential candidates for downstream development of novel drugs against intracellular bacterial infections.
IntroductionInfectious diseases pose an increasing critical global threat to both human and animal populations. A recent study estimated 4.71 million deaths associated with antibacterial-resistant infections annually and forecasted over 39 million deaths from antibiotic-resistant infections by 2050 (Collaborators and G.B.D.A.R (2024)). Even nowadays, many infections are difficult to treat, resulting in high-dose administration of antibacterials, unbearable antibiotic toxicity, delays in effective treatment, and increased mortality due to multidrug-resistant (MDR) infections (Kamaruzzaman et al., 2017).
Importantly, this challenge is specifically prominent in the context of infections caused by intracellular bacterial pathogens. These bacteria, including the genera Salmonella, Listeria, Brucella, Rickettsia, Legionella, Chlamydia, and Mycobacterium, invade host cells to establish an adapted replication niche, which facilitates their survival and dissemination. The intracellular lifestyle of these bacteria offers protection from the host’s humoral immunity, sequestration from neutrophils, and access to nutrients that may be scarce extracellularly (Petit and Lebreton, 2022; Li et al., 2023). As a result, intracellular bacterial pathogens have evolved to manipulate host cells to access their preferred niches within targeted cells. After invasion, bacteria are contained within a plasma membrane-derived vacuole, such as phagosomes or endosomes. Vacuolar bacteria, like Salmonella enterica (S. enterica) or Mycobacterium tuberculosis (M. tuberculosis), remain within the modified vacuoles, while cytosolic bacteria, like Listeria monocytogenes (L. monocytogenes), rupture the vacuole and reside within the host cytosol (Johnson et al., 2018; Gal-Mor, 2019; Petit and Lebreton, 2022; Li et al., 2023).
Intracellular pathogens have co-evolved with hosts and developed a striking ability to manipulate and subvert multiple host functions and pathways to facilitate their survival and transmission. In general, the process by which intracellular bacteria hijack host cells can be divided into four distinct stages: adhesion, internalization, survival/proliferation, and dissemination. Interfering with each one of these steps can help in controlling the infection and may be considered as a desired drug target.
Salmonella enterica, a ubiquitous foodborne pathogen, employs a sophisticated multistage strategy for host colonization and systemic dissemination, critically orchestrated by its type III secretion systems (T3SSs). Initial infection hinges on adhesion to host epithelial cells, followed by a rapid invasion of these non-phagocytic cells, a hallmark of Salmonella’s pathogenesis. This internalization is largely driven by the Salmonella pathogenicity island (SPI) 1-encoded T3SS, which translocates effector proteins to manipulate host actin dynamics, resulting in membrane ruffling and bacterial uptake (Fattinger et al., 2021). Once inside the cell, Salmonella establishes an intracellular niche, known as the Salmonella-containing vacuole (SCV), where it undergoes intracellular replication. This crucial step is facilitated by the SPI-2-encoded T3SS, which delivers a different array of effectors that remodel the SCV, prevent lysosomal fusion, and acquire nutrients, allowing the bacteria to survive and multiply within macrophages and other phagocytic cells (Liss and Hensel, 2015). Ultimately, Salmonella can achieve dissemination to systemic sites, including the spleen and liver, often within these infected phagocytes, further contributing to systemic disease (Gal-Mor, 2019).
Although significant progress has been made during the last decades in our understanding of how intracellular pathogens interact with their hosts (Kellermann et al., 2021), this group of pathogens remains a major clinical concern worldwide, and the gained knowledge has not been translated into new therapeutic approaches yet. This challenge has become even more urgent with the increasing prevalence of MDR strains that are often linked to a more severe disease outcome, and more strains that are resistant continue to emerge worldwide (Alara and Alara, 2024). Alarmingly, some of these bacteria are responsible for clinically important and prevalent diseases, including tuberculosis (TB), chlamydial infections, listeriosis, and invasive salmonellosis (Liu et al., 2020). Hence, there is a pressing need to better understand how intracellular pathogens interact with their hosts and to develop new approaches to treat these infections.
A novel anti-infectious therapeutic approach called host-directed therapy (HDT) is based on the modulation of host response by pharmacologically active compounds and has recently been highlighted as a promising strategy to counteract the emergence of antimicrobial resistance. This therapeutic approach aims to augment protective innate or adaptive host response needed for pathogen control and/or to limit its pathogenicity (Thom and D’elia, 2024). Because traditional antibiotics directly target bacterial processes, they employ strong selective pressure, driving the evolution of antibiotic resistance mechanisms in the pathogen. In contrast, HDT bypasses the pathogen’s direct resistance mechanisms, creating a favorable strategy to combat existing drug-resistant strains and slow the emergence of new resistance. However, the inherent complexity of host biology and immune response makes identifying effective, broad-ranged, and safe modulators challenging, unpredictable, and vastly complicated.
Traditional high-throughput screening (HTS) campaigns, largely designed to identify direct antibacterial agents in axenic culture, often fail to address the distinct challenges of intracellular pathogens, such as their shielded intracellular niche and complex host–pathogen interactions. Existing cell-based screens, while valuable, frequently rely on endpoint assays or indirect readouts that lack the sensitivity, specificity, and throughput required to robustly identify compounds that selectively inhibit intracellular bacterial replication or modulate host responses without direct cytotoxicity.
Here, we used the clinically relevant Gram-negative pathogen S. Typhimurium as a prototypic model for facultative intracellular bacterial pathogens and developed a cell-based HTS protocol, coupled with an intracellular lux-based reporter system, which allowed effective screening of a small molecule library comprising nearly 37,000 compounds. Eight non-cytotoxic compounds [saracatinib (AZD0530), penbutolol, AW00794SC, PCM-0103431, Arry-380 analog, conivaptan, SPB03336SC, and indacaterol maleate] with no growth inhibition in culture were found to efficiently inhibit the intracellular growth of S. Typhimurium in host cells. Interestingly, seven of these molecules were also demonstrated to inhibit the intracellular growth of the Gram-positive pathogen L. monocytogenes. Moreover, the structure–activity relationship (SAR) approach was applied for one leading hit, and chemical analogs with even improved infection-inhibiting activity were identified. Overall, we describe a robust HTS platform that can be adapted for the screening of compound libraries against other intracellular bacteria and parasites, and suggest that the identified compounds can be used as potential candidates for the development of novel drugs to treat intracellular bacterial infections.
Materials and methodsBacterial strains and growth conditionsAll strains and plasmids used in this study are listed in Supplementary Table 1. Salmonella enterica serovar Typhimurium strain SL1344 and Listeria monocytogenes strain EDG were used as prototypical Gram-negative and Gram-positive intracellular bacteria, respectively. Salmonella and Escherichia coli strains carrying the Pssek3::lux and PrpoD::lux (see Supplementary Table 1) were grown in LB-Lennox broth, PCN minimal medium (Neidhardt et al., 1974) [80 mM of MOPS pH 7.4/80 mM of MES pH 5.8, 1/0.4 mM of phosphate, 10 mM of glucose, 4 mM of tricine, 100 μM of FeCl3, 376 μM of K2SO4, 50 mM of NaCl, 10 nM of Na2MoO4, 10 nM of NaSeO3, 4 nM of H3BO3, 0.3 mM of CoCl2, 0.1 mM of CuSO4, 0.8 mM of MnCl2, 0.1 mM of ZnSO4, 15 mM of NH4Cl, 1 mM of MgSO4, 10 μM of CaCl2] with histidine, or in N-minimal medium [80 mM of MES (pH 5.8), 5 mM of KCl, 7.5 mM of (NH4)SO4, 0.5 mM of K2SO4, 337 μM of K2HPO4/KH2PO4 (pH 7.4), 20 mM of MgCl2, 38 mM of glycerol, and 0.1% casamino acids] with aeration at 37°C supplemented with kanamycin (50 µg/mL) with shaking (250 rpm).
Construction of bioluminescence reportersTo construct a bioluminescence reporter strain, the S. Typhimurium sseK3 promoter region was PCR-amplified using the primers “F XhoI sseK3 Pro” and “R BamHI sseK3 Pro” (Supplementary Table 2). The obtained 221-bp fragment was then cloned using the restriction enzymes XhoI and BamHI into the pCS26 vector that carries the luxCDABE operon of the bacterium Photorhabdus luminescens (Bjarnason et al., 2003; Aviv and Gal-Mor, 2018). The resulting construct, Pssek3::lux, was electroporated into S. Typhimurium SL1344-electrocompetent cells. Salmonella Typhimurium SL1344 and E. coli DH5 harboring the lux reporting constructs were grown in LB at 37°C with shaking (250 rpm) for 16 h and then washed and subcultured (1:100) into fresh LB or N-minimal medium. Growth curves of the reporter strains were obtained by growing the cultures in LB and N-minimal medium in a white 96-well flat-bottom plate (Greiner Bio-One, Kremsmünster, Austria) at 37°C under regular shaking. Absorbance at 600 nm and bioluminescence were determined using the Infinite 200 PRO M-Plex microplate reader (Tecan, Männedorf, Switzerland) and imaged using IVIS Lumina LT (PerkinElmer, Shelton, USA) after 5.5 h of growth.
Bioluminescence inhibition assayTo evaluate whether the compounds influence the bioluminescence of the intracellular reporter, luminescent S. Typhimurium was added to a sterile 96-well, cell culture-treated, flat-bottom white microplate, containing the compounds. An overnight culture of S. Typhimurium expressing PsseK3::lux was grown in PCN medium pH 7.4 with 1 mM of phosphate (non-luminescence-inducing medium) at 37°C, on a shaker incubator. Cells were sedimented at 13,000 rpm and washed with PCN medium pH 5.8 containing 0.4 mM of phosphate (luminescence-inducing medium) and used to inoculate a subculture 1:50 in PCN pH 5.8 with 0.4 mM of phosphate, grown for 6 h at 37°C on a shaker incubator. The subculture was diluted 1:20 with PBS, and luminescence induction was verified using the Tecan Infinite 200 PRO microplate reader. A total of 200 µL of the luminescent bacterial suspension was then added to each well of the microplate containing the compounds, and luminescence was measured directly, after 10 and 20 min. Bacterial suspension from a subculture grown in a non-luminescence-inducing medium was used as a reference control.
Antibacterial assaysOvernight cultures of S. Typhimurium and L. monocytogenes were subcultured 1:100 in liquid LB and BHI medium, respectively, in a 96-well microplate in the presence and absence of the tested inhibitors and at a final concentration of 10 µM. Bacterial growth was followed for 24 h at 37°C with regular shaking by optical density reading at 600 nm using the Infinite 200 PRO M-Plex microplate reader (Tecan).
Quantitative reverse transcription real-time PCRSalmonella Typhimurium SL1344 was subcultured 1:100 into fresh LB medium in the presence of 10 µM compounds C2 (PCM-0086166) and C3 (PCM-0004846) and grown for 3 h to the late logarithmic phase (OD600 ~ 1.0). RNA was fixed using the RNAprotect Bacteria Reagent (QIAGEN) and extracted using the RNeasy Mini Kit (QIAGEN), according to the manufacturer’s instructions. Quantitative reverse transcription real-time PCR (qRT-PCR) was conducted as previously described (Elhadad et al., 2016), with a few modifications. Briefly, cDNA was synthesized using the qScript cDNA Synthesis Kit (Quantabio) using a T100 thermal cycler (Bio-Rad). Each reaction in a final volume of 20 μL was conducted in a 96-well optical reaction plate (Applied Biosystems) containing 10 μL of FastStart Universal SYBR green Master mix, 2 μL of cDNA, and target gene-specific primers (Supplementary Table 2) at a final concentration of 0.3 μM each. Melting curve analysis was applied to verify that each qRT-PCR reaction has generated a single amplimer. Relative quantification of transcripts was determined using the comparative threshold cycle (CT) method (Livak and Schmittgen, 2001), while transcript levels were normalized to the housekeeping gene rpoD. The ΔCT values were calculated by determining the difference in threshold values for the target gene and rpoD in cultures grown in the presence of compounds versus the threshold values of cultures grown in the absence of compounds (LB broth). The ΔΔCT value was calculated by subtracting the normalized ΔCT value obtained for the LB culture from the normalized ΔCT value obtained for the culture grown with the compounds.
Small molecule libraryA total of 36,855 compounds (Supplementary Table 3) from the Weizmann Institute screening collection were included in this study. The compounds were compiled from several commercial screening collections (Sigma LOPAC, Prestwick Known Drugs, MicroSource-Spectrum, Selleck Bioactive, Maybridge HitFinder, Enamine, and ChemDiv). Compounds were dissolved to 10 mM stocks in dimethyl sulfoxide (DMSO) and stored in a desiccated nitrogen-flushed environment until use.
High-throughput screeningThe molecule library screen was performed in sterile tissue culture-treated white 384 flat-bottom well plates (Greiner) preloaded with 36,855 small molecules, at a final concentration of 10 µM using the Echo555 acoustic transfer system (Labcyte, Germany). Plates were seeded with HeLa cells suspended in DMEM containing 10% FCS (8 × 103 cells/well) using the Multidrop Combi Reagent Dispenser (Thermo Fisher Scientific, Waltham, USA) and incubated at 37°C in 5% CO2 humidified incubator for 4 h to allow the cells to adhere. Each plate included 16 wells of 2 μM of cytochalasin D (CD) that was used as a positive control. An overnight culture of S. Typhimurium expressing PsseK3::lux was subcultured 1:100 in LB medium and grown for 3 h at 37°C with shaking (250 rpm) to the late logarithmic phase (OD600 ~ 1). The cultures were then diluted 1:50, and aliquots of 25 µL were used to infect the cells at a multiplicity of infection (MOI) of 1:50 (cell to bacteria). One hour post-infection (hpi), gentamicin at a final concentration of 20 µg/mL was added to each well to eliminate extracellular bacteria, and the plates were incubated overnight (˜16 h) at 37°C under 5% CO2 atmosphere in a LiCONiC tissue culture incubator (LiCONiC Instruments, North Reading, USA). The next day, the infected cells were washed with phosphate-buffered saline (PBS), using a BioTek EL406 washer dispenser (BioTek Instruments, Winooski, USA), and the bioluminescence of the cells was detected by a LUMIstar Omega Plate Reader (BMG Labtech, Ortenberg, Germany), a rapid plate reader at detection mode: luminescence, a gain of 3,700, integration time of 200 ms, and plate chamber temperature of 37°C. Genedata Screener (Basel, Switzerland) was used for the normalization and interpretation of results.
Epithelial cell infection with Salmonella and ListeriaTo evaluate the infection inhibition activity of the compounds using direct bacterial counting, 5 × 104 HeLa cells were suspended in DMEM (+10% FCS), seeded in a 24-well, cell culture-treated, flat-bottom microplate (Greiner), and incubated at 37°C in a humidified 5% CO2 incubator overnight prior to infection. The following day, the medium was replaced with fresh DMEM (+10% FCS) medium containing the PCM-0094889, PCM-0086166, PCM-0004846, PCM-0103431, PCM-0095494, PCM-0095293, PCM-0001349, and PCM-0095564 compounds at a final concentration of 10 µM, which were incubated with the cells for 4 h before the infection. An overnight S. Typhimurium SL1344 culture was subcultured 1:100 into fresh LB medium and grown at 37°C on a shaker incubator to the late logarithmic phase. After 3 h, the bacteria were diluted 1:100 in DMEM, and 500 µL from the diluted bacteria were added into each well at an MOI of 1:50. The infected cells were spun down for 5 min at 1,000g and then incubated at 37°C under 5% CO2 atmosphere. After 10 min, cells were washed three times with PBS, and fresh DMEM medium supplemented with 10 µM of each compound was added to each well and incubated for an additional 20 min. At 30 min post-infection, the medium was replaced with a fresh medium containing 10 µM of each compound and 100 µg/mL of gentamicin and incubated for 90 min. At 2 hpi, cells were washed three times with PBS and lysed with 250 µL of lysis buffer (0.1% SDS, 1% Triton X-100 in PBS) by incubating the cells for 10 min at room temperature with gentle agitation. To calculate the number of invading bacteria at 2 hpi, the cell lysates were serially diluted in PBS and plated onto LB agar plates for CFU count. To calculate the intracellular fold replication, at 2 hpi, the medium of the infected cells was replaced with DMEM supplemented with 10 µM of each compound and 10 µg/mL of gentamicin and incubated at 37°C in a humidified 5% CO2 incubator. At 24 hpi, the cells were washed three times with PBS and lysed with 250 µL of lysis buffer as above. Intracellular replication was determined by the ratio between the number of intracellular bacteria (CFU) counted at 24 hpi and their number at 2 hpi.
To infect HeLa cells with L. monocytogenes, cells were prepared the same way and incubated for 3 h with fresh DMEM (+10% FCS) supplemented with the tested compounds at a final concentration of 10 µM. Overnight cultures of L. monocytogenes that were grown statically in BHI at 30°C were washed and resuspended in PBS. Cell infection at an MOI of 1:50 was achieved by adding 20 µL of Listeria suspension to each well, followed by incubation at 37°C under 5% CO2 atmosphere. Thirty minutes post-infection, the cells were washed with PBS, and the medium was replaced with fresh medium supplemented with 10 µM of the tested compounds. One hpi, gentamicin was added to each well at a final concentration of 50 µg/mL. At 6 hpi, the cells were washed with PBS and lysed with 250 µL of DDW at room temperature. The cell lysates were then diluted in PBS and plated on LB-agar plates for CFU count. Listeria infection was calculated as the number of intracellular bacteria recovered at 6 hpi, divided by the infecting inoculum (CFUs).
For infection of HT29 cells, 3 × 105 HT29-MTX-E12 cells per well were seeded in a 24-well plate. The day after seeding, cells were treated with the inhibitors at the indicated concentrations. Three hours later, cells were infected with a mid-logarithmic culture of S. Typhimurium SL1344 at an MOI of 30. After 30 min, cells were washed three times, and medium containing inhibitors and gentamicin (100 µg/mL) was added. One hour later, the medium was removed and replaced with the medium containing inhibitors and gentamicin (10 µg/mL). Twenty-four hpi, cells were washed three times with PBS and lysed with 500 µL of lysis buffer (0.1% SDS, 1% Triton X-100 in PBS) for 10 min at room temperature. Serial dilutions were plated on LB agar plates for CFU counting.
Dose–response relationship assay in HeLa cellsTo evaluate the inhibition activity of the compounds in a dose–response assay, 4.6 × 104 HeLa cells were seeded in each well of a sterile 96-well, cell culture-treated, flat-bottom white microplate. Cells were incubated at 37°C/5% CO2 humidified incubator for 4 h in the presence of 0, 0.25, 1, 3.25, 10, and 30 µM of compounds PCM-0094889, PCM-0086166, PCM-0004846, PCM-0103431, PCM-0095494, PCM-0095293, PCM-0001349, and PCM-0095564. An overnight S. Typhimurium culture expressing Pssek3::lux was subcultured 1:100 in LB medium for 3 h at 37°C on a shaker incubator to the late logarithmic phase (OD600 ~ 1). The Salmonella culture was then diluted 1:50 in DMEM, and aliquots of 100 µL were added to each well to reach an MOI of 1:30 (cells per bacteria) and incubated at 37°C, 5% CO2 to allow infection. One hpi, gentamicin was added to a final concentration of 20 µg/mL, and the plates were incubated overnight. The next day, the cells were washed three times with sterile PBS, and bioluminescence was read using the Infinite M-Plex Tecan plate reader.
Bone marrow-derived macrophage infectionBone marrow-derived macrophages (BMDMs) were isolated from the femur leg bone of 7-week-old SWISS female mice. Macrophages were diluted in BMDM medium (50% DMEM high glucose, 20% FCS, 30% L-929 conditioned medium, 2 mM of L-glutamine, 1 mM of sodium pyruvate, 50 nM of β-mercaptoethanol) and seeded at 2.5 × 105 cells/ml in a 24-well, cell culture-treated dish, 24 h prior to Salmonella infection. The next day, the BMDM medium was replaced with fresh BMDM medium supplemented with 10 µM of the tested compounds and incubated for 3 h at 37°C under 5% CO2 atmosphere. Macrophages were infected at an MOI of 1:10 with cultures of S. Typhimurium SL1344 and its ΔssaR isogenic mutant as a negative control. Infected cells were spun down at 1,000g for 5 min and incubated for 30 min at 37°C, 5% CO2. Then, cells were washed three times with PBS to remove extracellular bacteria, and BMDM medium containing 100 μg/mL of gentamicin was added for 1 h of incubation. Cells were then washed three times with PBS, and the medium was replaced with fresh BMDM containing 10 μg/mL of gentamicin. To determine the intracellular growth of Salmonella at 2 and 24 hpi, cells were washed three times with PBS and lysed with 250 μL of lysis buffer (0.1% SDS, 1% Triton X-100 in PBS). The number of intracellular CFUs was quantified by plating serial dilutions of cell lysates on selective (streptomycin) LB-agar plates. Salmonella uptake was calculated as the number of intracellular bacteria recovered at 2 hpi, divided by the infecting inoculum (CFUs). Salmonella survival was calculated as the number of intracellular bacteria recovered at 24 hpi, divided by the number of intracellular bacteria recovered at 2 hpi.
Cytotoxicity assayTo examine the potential toxicity of the identified hits to host cells, the CellTiter-Glo (GTG) Luminescent Cell Viability Assay (Promega, Madison, USA) was used. Sterile tissue culture-treated white 384 flat-bottom well plates (Greiner) were loaded with compounds at final concentrations of 0.3, 1, 3, 10, and 30 µM using the Echo555 acoustic transfer system (Labcyte, Germany). HeLa (2 × 103) or HB2 epithelial cells were suspended in DMEM containing 10% FCS, and 50 µL of cell suspension was added to each well using Multidrop (Thermo Fisher Scientific) and incubated for 5 h. Gentamicin at a final concentration of 20 µg/mL was added to each well, and the cells were incubated overnight. The CTG assay was performed in accordance with the manufacturer’s protocol. Briefly, the medium was aspirated, and 10 µL of the CTG reagent was added to each well. Then, the plates were shaken for 2 min followed by 10 min of incubation at 37°C under 5% CO2 atmosphere. Luminescent signal was detected using a PheraStar FS plate reader (BMG Labtech, Ortenberg, Germany). The results were analyzed using GeneData software (Basel, Switzerland).
SAR studiesTo implement a SAR study (Wawer and Bajorath, 2009), 41 commercially available close analogs of compound C4 (Supplementary Table 5) were obtained from Molport EU (www.molport.com) and tested for their ability to inhibit S. Typhimurium infection of human epithelial cells. To test the activity of the chemical analogs, 4.6 × 104 HeLa cells were seeded in each well of a 96-well, cell culture-treated, flat-bottom white microplate (Greiner) and incubated at 37°C in 5% CO2 humidified incubator for 4 h, in the presence of 10 µM of compound PCM-0103431 and its chemical derivatives (Supplementary Table 4). Cells were infected with a subculture of S. Typhimurium expressing the reporter system Pssek3::lux as described above at an MOI of 1:50. One hpi, gentamicin was added to a final concentration of 20 µg/mL, and the infected cells were incubated overnight. The next day, the wells were washed three times with PBS, and bioluminescence was read using the Infinite 200 PRO M-Plex microplate reader (TECAN).
ResultsConstruction and functional characterization of a reporter system for intracellular Salmonella TyphimuriumTo facilitate efficient screening of molecules that can potentially inhibit host cell infection by S. Typhimurium, we sought to construct a reliable reporter system that could be used in a quantitative HTS. Since bioluminescence generally provides a broader dynamic range and lower noise-to-signal ratio over fluorescence (Fan and Wood, 2007), we decided to use a self-sufficient bacterial luminescence system that does not require the addition of an external substrate (Aviv and Gal-Mor, 2018). For that purpose, we cloned the S. Typhimurium rpoD and sseK3 promoters upstream to the luxCDABE operon of the bacterium Photorhabdus luminescens carried on the plasmid pCS26 (see Supplementary Table 1). RpoD (σ70) is the primary housekeeping sigma factor, responsible for the expression of Salmonella genes required for exponential growth under normal cellular conditions (Park et al., 2024), while SseK3 is a T3SS-2 translocated effector, whose expression is induced, while Salmonella is intracellular or under SPI-2-inducing conditions (Brown et al., 2011). Figure 1 shows the bioluminescence signal of these reporter strains during growth in liquid culture in LB broth (Figure 1A) and in acidic N-minimal medium (NMM; pH 5.8; Figure 1B), considered as SPI-1- and SPI-2-inducing conditions, respectively.

Functional characterization of a luminescence reporter system as a readout for intracellular Salmonella Typhimurium. Salmonella Typhimurium SL1344 harboring the empty autobioluminescent plasmid pCS26, PropD::lux, or PsseK3::lux and Escherichia coli DH5 harboring PropD::lux were subcultured into LB (A) or N-minimal medium (B) and grown for 6 h at 37°C under constant shaking. The optical density and the bioluminescence of the cultures were measured and expressed as the mean bioluminescence reading (of four replicates) normalized to their optical density at 600 nm (OD600). An image of the bioluminescence signal produced by these cultures was taken using the IVIS Lumina LT (PerkinElmer) imaging system and is shown after 5.5 h of growth from subculture. (C) HeLa cells were suspended in DMEM medium, seeded in white 96-well plate, and incubated at 37°C under 5% CO2 for 4 and 16 h prior to infection to allow the cells to adhere. Cells were then infected with E. coli expressing the PrpoD::lux and S. Typhimurium strains expressing either PrpoD::lux or Pssek3::lux. Non-internalized bacteria were killed by gentamicin, and at 16 h post-infection, bioluminescence was recorded and expressed in relative light units (RLU). Uninfected cells (mock) or cells infected with S. Typhimurium (STM) harboring the empty vector (pCS26) were used as negative controls.
While the S. Typhimurium strain expressing the PrpoD::lux reporter showed similar signal levels of luminescence in LB and NMM, the Pssek3::lux reporter was prominently induced in an S. Typhimurium strain grown in NMM and demonstrated high levels of luminescence under these conditions. Interestingly, an E. coli strain expressing the lux operon under an S. Typhimurium rpoD promoter also produced high levels of bioluminescence both in LB and NMM, indicating high and constitutive expression of this promoter in the E. coli background.
To test the ability of these strains to be used as a reporter system for intracellular bacteria, HeLa cells were infected with E. coli expressing the PrpoD::lux and S. Typhimurium strains expressing either the PrpoD::lux or Pssek3::lux reporters. Non-internalized bacteria were killed by the addition of gentamicin to the medium, and at 16 hpi, bioluminescence was examined as a readout for the intracellular bacterial load. HeLa cells infected with S. Typhimurium expressing the PrpoD::lux or Pssek3::lux constructs resulted in a high and specific bioluminescence signal that was readily detected in the infected host cells. Uninfected cells (mock) or cells infected with S. Typhimurium carrying the empty vector produced almost no luminescence, indicating a low background-to-signal ratio. Similarly, in contrast to the high signal obtained by the E. coli strain harboring the PrpoD::lux reporter extracellularly, this strain produced very low bioluminescence signal in the context of HeLa cell infection, due to its poor ability to invade these epithelial host cells (Figure 1C).
Because the signal of the PsseK3::lux reporter was higher than the signal of PrpoD::lux in HeLa cells infected with S. Typhimurium, and because similar results were obtained after 4 or 16 h of incubation host cells to allow sufficient cell adhesion prior to the infection, we decided to apply 4 h of incubation of host cells before infection with S. Typhimurium carrying the PsseK3::lux reporter in the subsequent HTS.
A cell-based HTS successfully identified Salmonella infection inhibitorsThe S. Typhimurium strain expressing the PsseK3::lux reporter was used in a seven-stage screening cascade, aiming to identify small molecules that can inhibit intracellular growth of S. Typhimurium. The different stages of the screening campaign and the number of compounds that passed each one of the filtering criteria are illustrated in Figure 2A and summarized in Table 1, respectively. To facilitate this HTS, HeLa cells were seeded in 384-well plates and incubated for 4 h in the presence of 36,855 compounds composing a collection of small molecules from different commercial libraries of putative bioactive molecules, at a final concentration of 10 µM (Supplementary Table 3). The incubated cells were then infected with the reporter S. Typhimurium PsseK3::lux strain for 1 h, washed, and incubated in the presence of gentamicin. Gentamicin is a potent bactericidal antibiotic that effectively kills extracellular Salmonella, but has poor penetration into eukaryotic host cells, and therefore, intracellular bacteria are “protected” from killing and survive this treatment. At 16 hpi, the bioluminescence signal in each well was determined by a plate reader and compared to the signal of cells that were incubated with 0.1% DMSO and infected with S. Typhimurium expressing PsseK3::lux, which was used as a reference control (Ctrl). As a positive control for a potent infection inhibitor, we included CD that blocks actin polymerization and therefore inhibits Salmonella invasion into host cells (Finlay et al., 1991). Since the intensity of the luminescence is generally proportional to the number of intracellular Salmonella, compounds that inhibit either the bacterial invasion or their intracellular replication are expected to result in a lower bioluminescence signal. Therefore, molecules that reduced the bioluminescence in the infected cells by 50% or more were selected as potential infection inhibitors for the next screening stage. During the first screening stage, we identified 473 (1.2% from all tested compounds) molecules that met this inclusion criterion.

Cell-based high-throughput screening and identification of host cell infection inhibitors. (A) A schematic illustration of the pipeline used for the HTS. Infection assay of the intracellular pathogen S. Typhimurium carrying the reporter system was performed in HeLa cells in the presence of 36,855 bioactive molecules. Compounds that were found to reduce the bioluminescence signal (N = 473) were selected for the next stage. To filter out compounds that are cytotoxic to HeLa cells, the compounds were examined for cytotoxicity in HeLa cells at 10 µM concentration. Non-cytotoxic compounds (N = 175) continued to the next screening step. The antibacterial activity of the compounds was tested, and 101 compounds that showed antibacterial activity against S. Typhimurium were filtered out. Seventy-four qualified compounds were tested in a second independent test to confirm their ability to inhibit Salmonella infection. Hits that presented at least 50% decrease in the intracellular bioluminescence signal (N = 68) were selected for the next stage. Six out of 68 compounds that were found unstable in a liquid chromatography–mass spectrometry (LC–MS) analysis were excluded, and the remaining 62 compounds were tested for their ability to inhibit Salmonella infection in the dose–response infection experiments. At the end of the screen, we identified eight molecules that were able to inhibit Salmonella infection in HeLa cells and showed low cytotoxicity, no antibacterial activity, and high purity. (B) The ability of the eight compounds (C1 to C8) to inhibit Salmonella Typhimurium (STM) invasion into HeLa cells was determined by the bioluminescence assay as described in the Materials and methods section. Salmonella infection in the presence of the eight compounds at a final concentration of 10 µM is shown relative to its infection in the absence of the compounds (Ctrl). Cytochalasin D (CD) that blocks Salmonella invasion was used as a positive control. The bars show the mean and the standard error of the mean (SEM) of three to six biological repeats. One-way ANOVA was used to determine statistical significance. ***, P-value < 0.001.
Screening stagePhenotype testedAssay usedInclusion criteriaNumber of tested moleculesNumber of qualified moleculesNumber of technical repeatsNumber of biological repeats1HeLa cell infectionBioluminescence signal of intracellular bacteria50% or more reduction in the bioluminescence intensity36,855473112Toxicity to HeLa cellCellTiter-Glo Luminescent Cell Viability Assay90% or more viability of HeLa cells following 16 h incubation473175123Antibacterial against SalmonellaBacterial growth in LB by OD600 readingNo significant inhibition of S. Typhimurium growth17574134HeLa cell infectionBioluminescence signal of intracellular bacteria50% or more reduction in bioluminescence intensity7468125Chemical stabilityCompounds purity by LC–MS analysisPurity of 70% or more6862116No effect on bioluminescence productionBioluminescence signal in bacterial cultureReduction of ≤30% in luminescence6254127HeLa cell infectionBioluminescence signal of intracellular bacteria in the presence of 0.25, 1, 3.25, 10, and 30 µM drugDose–response behavior54813Different stages of HTS to identify Salmonella infection inhibitors.
However, since a reduced signal can also result from cytotoxicity to HeLa cells or antibacterial activity against Salmonella, further filtering of candidates was applied in the next steps to eliminate compounds with an undesired cytotoxic mode of activity. To this end, we tested the cytotoxic activity of the 473 candidates at 10 µM concentration, using the CellTiter-Glo Luminescent Cell Viability Assay (Promega), and continued screening only compounds that maintained HeLa cell viability of 90% or more after incubation for 24 h in the presence of the compounds. Filtering out 298 compounds that did not meet this threshold resulted in 175 molecules that were qualified for the next screening stage.
In the following step, we sought to filter out compounds with antibacterial activity, as we wanted to avoid molecules that directly kill or inhibit bacterial growth, and focus only on those that can specifically inhibit host cell infection. To this end, we tested the ability of the 175 eligible compounds to inhibit the growth of subcultured S. Typhimurium in LB medium by following their optical density at 600 nm for 24 h. As expected, a significant portion of 101 out of the 175 compounds tested was found to impair Salmonella extracellular and cell-independent growth, and after their exclusion, we continued the screening with 74 non-cytotoxic molecules that showed no growth inhibition under the tested conditions. This group of molecules was tested again, in a second independent assay, for their ability to inhibit intracellular growth of S. Typhimurium, by following their bioluminescence signal in HeLa cells at 16 hpi. In this stage, 68 out of 74 tested compounds demonstrated again 50% or more reduction of the intracellular bioluminescence signal and were therefore selected for the next screening stage.
Subsequently, we tested the 68 selected compounds using liquid chromatography–mass spectrometry (LC–MS) to confirm the presence of >70% expected mass of these molecules. Six compounds that did not pass this criterion were excluded from the screen, resulting in a short list of 62 hits. Among this group of compounds, eight molecules were found to inhibit the bioluminescence production in S. Typhimurium culture that was grown in PCN pH 5.8 medium (Deiwick et al., 1999) as a ssek3-inducing medium (Supplementary Table 4) and, therefore, disqualified for downstream analysis. The remaining 54 compounds were next tested in a dose–response infection assay, using their bioluminescence as a readout for host cell infection. In these experiments, the ability of the 54 tested compounds to inhibit Salmonella intracellular growth in HeLa was studied under increasing drug concentrations of 0.25, 1, 3.25, 10, and 30 µM. Consequently, in this last stage of the screen, eight compounds (PCM-0094889, PCM-0086166, PCM-0004846, PCM-0103431, PCM-0095494, PCM-0095293, PCM-0001349, and PCM-0095564) that presented a clear dose–response behavior (Supplementary Figure 1A) were selected as the final candidates of the screen. For simplicity, these compounds will be called hereafter as C1 to C8 (Table 2). The inhibitory effect of these final eight compounds at 10 µM concentration on HeLa cell infection, by looking at the intracellular luminescence of the reporter S. Typhimurium strain, is shown in Figures 2B, and their cytotoxicity activity toward HeLa and HB2 epithelial cells is shown in Supplementary Figure 1B.
#PCM numberMolecule namePCM structureMWIntracellular luminescence (%) relative to mock ± SEHeLa viability (%)C1PCM-0094889Saracatinib (AZD0530)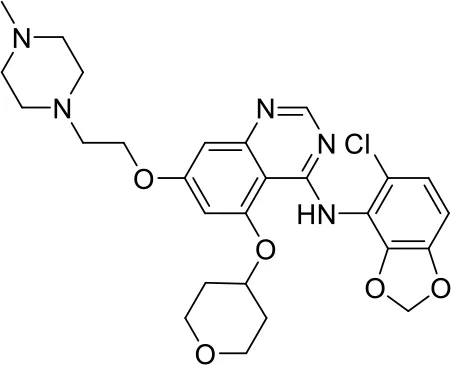 542.017 ± 496.0C2PCM-0086166Penbutolol
542.017 ± 496.0C2PCM-0086166Penbutolol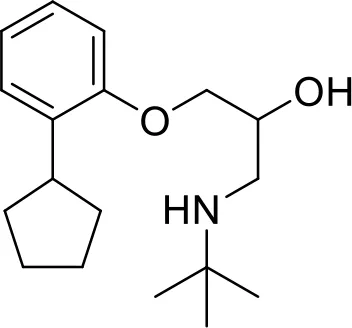
Comments (0)