
Remember me

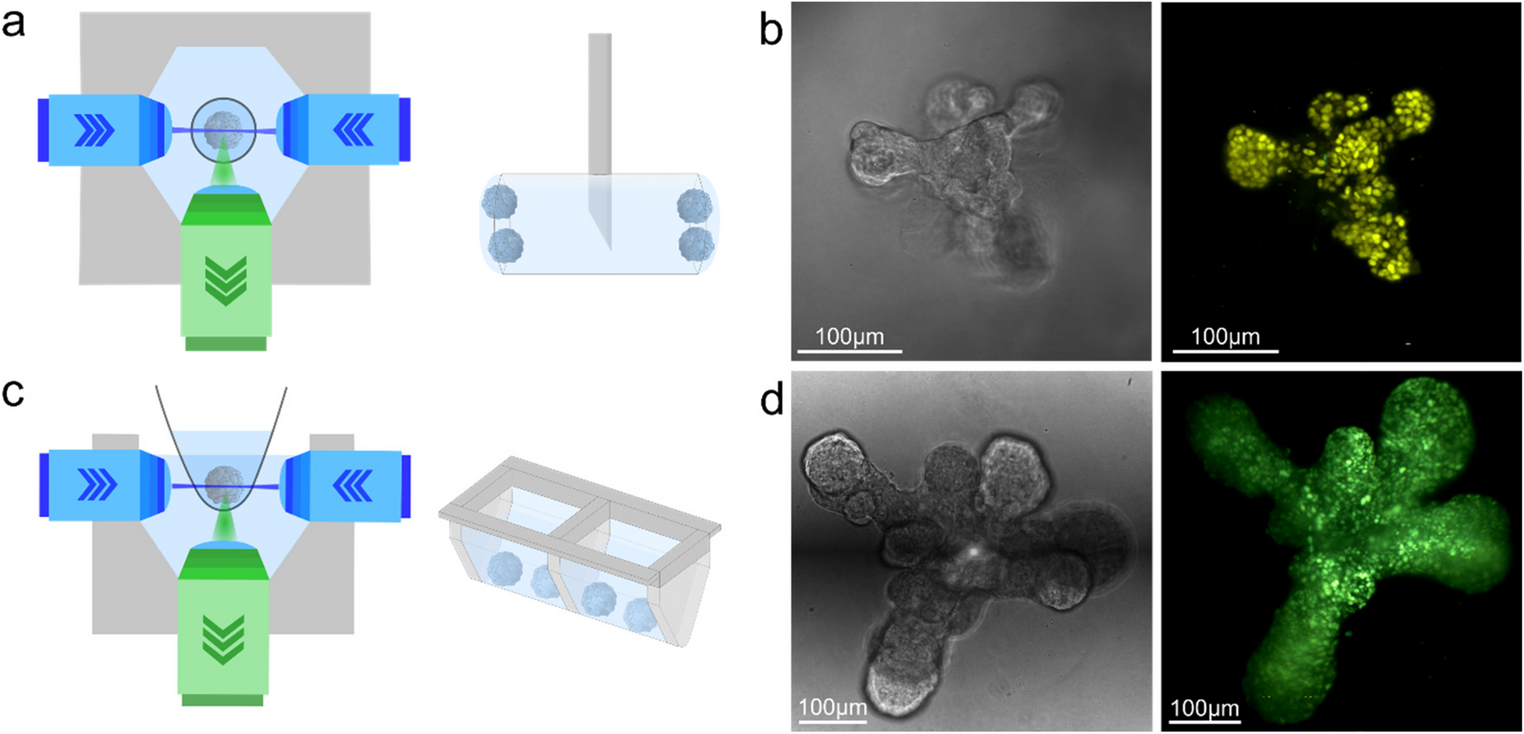



As an example of biogrowleR capabilities and illustrating the workflow, in this and the next section, we will use data on in vivo growth of a patient-derived xenograft (PDX) using bioluminescence measurements. The data were generated from slow growing estrogen receptor positive breast cancer samples using the intraductal xenografting approach (MIND) [9, 10]. These tumors grow slowly and the resulting data are challenging to analyze, as measurements are often amiss and the experimental groups are unbalanced. Estrogen receptor-positive (ER+) breast cancer is a hormone-driven disease treated with targeted therapies. Steroid hormones, such as estrogen and progesterone affect tumor cell proliferation. In order to better model ER + BC in vivo and understand the effects of estradiol (E2), progesterone (P4) and their combination on the tumor cells, PDXs were generated using the MIND approach. For this, tumor cells were engineered to express the Green Fluorescent Protein-luciferase (GFP-Luc) reporter. Luciferase is an enzyme that when in presence with the luciferin substrate leads to a light-emitting reaction, referred to as bioluminescence. Mice were then injected with patient derived GFP-Luc expressing tumor cells into the ducts of one or multiple mammary glands. Tumor cells expressing luciferase were tracked in the animals in vivo thanks to the cell bioluminescence in the presence of luciferin. When the average luminescence reached a defined threshold, mice were treated with vehicle, E2, P4 or combined E2 + P4 for 64 days by means of slow-release drug pellets implanted subcutaneously [10]. Thus, bioluminescence emanating from each individual mammary gland could be measured, and differences in signal over time and across the treatment conditions could be compared.
The visualization tools are readily available on R or any other programming language and recommendations are provided. The first step is to explore the data and to identify outliers. Outliers occur when expression of the GFP-luciferase reporter is shut down or the injection fails. Individual time plots can be viewed for each sample, i.e. each individual mouse mammary gland injected with labelled tumor cells (Fig. 2a). Such plots reveal different types of patterns of bioluminescence measurements over time and inform the modelling of the data. When plotting the individual growth curves together, stratified by different treatments, some control (CTRL) and P4 treated samples show a sharp drop in a single measurement around day 20 (Fig. 2b). This is likely attributable to an unsuccessful luciferin injection and could be considered as an outlier measurement. Raw images obtained from the in vivo imaging system (IVIS) measurements (Fig. 2c) may show samples without detectable signal. Stereomicroscopy complements IVIS images and reveals the distribution of fluorescence emitted by the GFP reporter in the mammary gland ducts. A well injected sample has fluorescence signal distributed all over the mammary gland reflecting cells that spread well within the milk ducts (Fig. 2d). On the other hand, a badly injected sample typically shows individual large foci, attributable to cells that are not well spread but growing outside the ducts (Fig. 2e).
Fig. 2
Steps of the exploratory data analysis. a Luminescence curves for each mouse and measurements taken. The color corresponds to the different treatment given to the mice for 64 days. b All growth curves for all glands plotted and stratified by condition. The curve in red corresponds to the average of all curves in the specified condition. The blue square in the P4 plot highlights an outlier measurement of one of the growth curves. c Representative IVIS image from mice with injected glands d Representative fluorescence stereomicrograph of an inguinal mammary gland in which the intraductally injected RFP expressing tumor cells spread within the mouse milk ducts. e Representative fluorescence stereomicrograph of an injected mammary gland where RFP expressing tumor cells have invaded into stroma the ducts. f Growth curves summarized in log10 or g Graph showing Total Flux (photons/second) over time of treatment. Error bars represent standard error
The ultimate goal of this type of analysis is to identify changes in total flux (photons/second) relative to a control condition. To this end, a summary growth curve shows the \(\:\text\text}_\)-scale total flux (Fig. 2f) and the total flux (Fig. 2g), for each condition. These curves illustrate some sources of variability and the influence of each measurement on the mean. For example, the measurements around day 20 for the CTRL samples were significant enough to lower the mean. Error bars highlight the extent of variability. Overall, in the present example there are no samples that can be considered outliers, so no measurements need to be discarded.
Modeling the Data Generating ProcessEach gland with injected tumor cells in a mouse corresponds to an experimental unit. Up to four glands in the same mouse are routinely injected. This experimental unit is the first level of nesting in the data since measurements are taken sequentially for each gland and so a timepoint is not independent from the previous one. Glands are further stratified by mice, being the second level of nesting. Mice can be housed in different cages, leading to a third level of nesting. Next, each mouse is assigned to a specific treatment, in this case the different hormone treatments. This is a covariate of interest. Another covariate of interest is time, i.e., the time at which the measurements were taken. To conclude, the \(\:\text\text}_\) total flux is modeled as a function of time and treatment covariates as fixed effects, and the repeated measurements for a single gland are included as a grouping variable in the hierarchical model. Equation 1 represents the R formula used to estimate the effects of time and treatment considering repeated measurements.
$$\begin\:\text\text}_\left(\text\text\text\text\text\:\text\text\text\text\left(\frac\text\text\text\text\text\text}\text\text}\right)\right)=&\;\text\text\text\text+\text\text\text\text\text\text\text\text\text+\text\text\text\text\\&\times\text\text\text\text\text\text\text\text\text+\left(1|\text\text\right)\end$$
(1)
Each condition has a different starting average total flux (Fig. 2a and b) captured by \(\:\left(1\:\right|\:id)\) in the formula (Eq. 1). Equation (1) is independent of the statistical framework used and both the frequentist or the Bayesian approach can be applied as described below. This proposed approach is called hierarchical modelling, described in more detail below, or mixed/random effects models.
Bayesian FrameworkThe Bayesian framework focuses on the interpretation of the results based on the posterior distribution of estimated parameters from the data. The posterior distribution is proportional to the likelihood of the collected data multiplied by the prior information available (Eq. (2), Bayes theorem), for example past events or previous experiments.
$$\:\text\text\text\text\text\text\text\text\text\propto\:\text\text\text\text\text\text\text\text\text\text\:\times\:\text\text\text\text\text$$
(2)
The posterior distribution can also be expressed in terms of condition probabilities (Eq. (3)):
$$\:\text\left(\text|\text\right)=\frac\left(\text|\text\right)\text\left(\text\right)}\left(\text\right)}$$
(3)
where A and B are events and P(B) is different from 0. The probability of event A happening given event B (P(A|B)) is a conditional probability also called the posterior probability. The P(B|A) is the probability of the event B given A, or likelihood of the data if A corresponds to the data and B are the parameters being estimated when performing Bayesian inference. P(A) and P(B) correspond to the prior information without any conditions. Since parameters are distributions, probabilistic arguments can be made.
Assuming that the model captures the data generating process, different probabilistic arguments can be used to explain the effect size. In our example, the bioluminescence signals of each PDX over time are modeled by linear regression using the mouse as the grouping variable (corresponding to each individual unit of measurement). In this way, growth rate estimates are obtained for each treatment (Fig. 3b) and comparisons between conditions can be made. One of the differences between Bayesian and frequentist frameworks concerns the uncertainty intervals. On the one hand, in the frequentist framework, the confidence interval assumes that data can be sampled an infinite number of times and the true estimate, in this case the growth rate, will be within the confidence interval 95% of the time. On the other hand, the uncertainty interval associated with estimates obtained by the Bayesian approach represents the credible interval. Bayesian estimates are based on the posterior distribution, which describes the uncertainty about statistical parameters conditional on a collection of observed data. In the example, based on the linear regression estimates, growth rates can be compared across conditions (see Methods) (Fig. 3c). In this context, the initial question “is there any difference between the conditions” can be resolved by rephrasing it to “what is the probability that the effect size is greater than 0”. This is different from the frequentist point of view that depends on a null hypothesis. Since we are modelling the data generating process, there is no null hypothesis. Figure 3c shows the effect size distributions for each pairwise comparison of interest. Each dot corresponds to a 1% quantile of the posterior distribution of the difference in growth rates. Thus, to answer the initial question, we have to determine the probability that the effect size is greater than 0. It is necessary to count the number of dots that exceed 0. When comparing E2 versus CTRL, 98 of the 100 dots are above 0 indicating a 98% probability this is the case. There is no established probability threshold for considering a significant effect because the question can always be reformulated, and the probability will change in each case. The next step is to determine whether there is any difference between conditions of interest. Rather than tackling this problem directly, it may be more useful to understand the magnitude and direction of the differences. This is done by modeling the data generating process through the use of statistical models (Fig. 3a). After choosing the correct statistical model, or the one closest to the data generating process, the model needs to be validated through convergence checks and other methods [1]. Problems in model validation can result from misspecification, i.e., the model does not match the data generating process, there is an insufficient number of data points, or from too many outliers. Once a valid model is obtained, it can be compared with a baseline model and inferences can be made, and the effect size can be determined. In the case of the growth curves different features can be analyzed; first, the difference between the slopes of the growth curves when modeling the data with linear regression and corresponds to the interaction term, second the difference between the initial and final values for two different conditions. The last option does not depend on linear models and polynomial or splines can be used to model the data.
Fig. 3
Modeling and interpreting the results. a Scheme showing the proposed workflow comprising data generation process and interpretation of the results. b Growth curves with overlapping best-fit line estimates from a hierarchical linear regression using each individual measurement with varying intercept. The different shades of the interval represent the 25% and 50% uncertainty intervals obtained from the posterior distribution. c Dot plots showing the distribution of effect sizes for each individual treatment comparison. Each dot corresponds to a 1% quantile of the expected posterior distribution. The effect size was calculated as the difference between the slopes of the estimated lines using all the time points. d Differences of the predicted growth curves when compared either to control or E2. CTRL: control, E2: 17-β-estradiol, P4: progesterone, E2P4: 17-β-estradiol plus progesterone. e Interaction terms obtained when using the frequentist approach for all pairwise comparisons
It is also important to consider the growth kinetics and/or the shape of the curves to understand to what extent conditions, in our case hormone treatments, increase or decrease growth. Figure 3d shows an alternative way of looking at the difference between some of the pairwise comparisons of interest. The direction of estimated growth gives an indication of the differences between the conditions of interest. The different starting points of the curves show that some of the conditions had a higher or lower total flux at the beginning of the experiment. Informative priors are encouraged in order to stabilize the parameter estimation (see Methods: Hierarchical Modelling). For example, in the particular MIND-PDX experiments, the prior distribution is a normal distribution with mean 7 and a standard deviation of 2. The mean 7 corresponds to an average value of \(\:lo_\) total flux usually used as a threshold to start a drug treatment in engrafted tumor experiments [9, 10].
Frequentist FrameworkAccording to the frequentist approach, the likelihood ratio test can be used to compare different models and thereby to determine whether growth rates differ between conditions. The idea is to have one full model that includes the interaction between time and treatment and another model in which this interaction is not present. The interaction estimates the differences between growth rates of different treatments and assumes the difference between the growth rates of the treatments is equal to 0 (null hypothesis), while the alternative hypothesis is the difference is not 0. To evaluate the null hypothesis we use the lrtest function from the R package lmtest to obtain a final p-value. If the question of interest concerns only two treatments, the models must be fitted to a subset of the data containing only the conditions of interest. Again, the difference between the fitted lines can be calculated. Confidence intervals for the estimated differences are calculated using bootstrapping [11].
In our example, Table 1 shows the results of the different statistical tests performed. The results obtained are similar to the Bayesian ones. Only the interpretation is different. With a significance level \(\:=0.05\), the null hypothesis for E2 vs. CTRL and E2 vs. E2 + P4 is rejected with E2 having higher growth rates in both comparisons. The interaction term and its 95% confidence interval can also be visualized (Fig. 3e). The interaction term corresponds to the difference in growth rate without any adjustment for initial variance at the beginning of the treatment. To summarize, these are the two ways to analyze and interpret growth measurement data once the data generating process is modeled.
Table 1 Results of the likelihood ratio test (LRT) for each pairwise comparison are given in the Comparison column. The p-value column indicates the p-value obtained from the LRT and the associated test statistics are provided in the Statistics columnSample RandomizationWhen using in vivo models, it is important to understand growth kinetics to inform the appropriate starting point for any treatments. Overall growth kinetics should be considered when assigning mice to the different treatment cohorts due to inherent variability. Longitudinal measurements are acquired prior to treatment to use for randomization. We developed a new algorithm to randomize mice into different groups, considering the growth kinetics or the shape of the growth and, if necessary, blocking factors, such as caging, to avoid further bias.
First, data are explored to detect outliers (Fig. 4a). All 4 injected glands of mouse 13 show constant signal, while luminescence increases exponentially with time in all other injected glands. This suggests that the tumor cells did not engraft well in the glands in mouse 13. As such, these glands need to be excluded from randomization to prevent skewing of the data.
Fig. 4
Randomizing and defining groups. Steps in the randomization process. a Plots showing in vivo luminescence of individual xenografted glands of three host mice prior to treatment. b-e Metrics used for randomization as a function of the total number of iterations used by the randomization algorithm. Four different metrics are used: i) pairwise difference of the average endpoint measurements for the expected group, ii) pairwise difference of the slopes for the assigned groups in each iteration, and respectively the same for iii) standard error of the intercept and iv) the standard error of the slopes. f Average growth curves for the two groups assigned by the randomization algorithm. The x-axis shows days before treatment in decreasing order
Subsequently, the retained measurements are randomized. In the case of in vivo tumor measurements, there may be multiple measurements per mouse. Therefore, it is important to make sure that in a single mouse the measurements are not highly variable. In this case, it is recommended to remove the entire experimental unit from the randomization process so as not to distort the grouping. Randomization is then performed at the level of individual mice. In the present example, we randomize 8 mice into two groups, named cluster 1 and 2. The method seeks to minimize the sum of pairwise differences of: growth rates, estimated averages of last luminescence measurements, standard deviation of growth rate, and standard deviation of the estimated average of last luminescence measurements between the groups. Since pairwise differences are considered, the number of groups is limited only by the number of available experimental units. By default, the get_best_splitting function runs for 100 iterations minimizing the mentioned metrics (see Methods: Simulated annealing). “Diagnostic plots” allow to check convergence of the minimization process and to control successful randomization. The convergence curves (Fig. 4b-e) show the value of the respective metrics in each iteration. As a general rule, the sum of pairwise differences at the last measurement point and of the growth rates should decrease with iterations while the sum of pairwise differences between standard deviations should be stable. An acceptable situation is a decrease in the summed pairwise difference between the last measurement point of the groups (Fig. 4b) and increase in the summed pairwise difference of the growth rates (Fig. 4c). The final grouping can be extracted from the results of the get_best_splitting function, and the growth curves can be visualized using the default functions in biogrowleR (Fig. 4f).
Following randomization it is important to ensure that groups are balanced with regards to other factors that may influence long-term measurements like cages and weight. Our algorithm includes a way to randomize mice into groups by blocking any categorical factors.
Comments (0)