
Remember me

Linear discriminant analysis is a classification method that calculates the probability that a multidimensional data point belongs to one of the user-defined classes. The algorithm, trained with data points belonging to known classes, finds a combination of dimensions that minimizes the variance within each class and maximizes the variance between classes (Balakrishnama and Ganapathiraju 1998; Tharwat et al. 2017). In our previous work (Romero et al. 2022), we used LDA to find the most differentiating combination of chemical shifts, allowing us to assign a new spin system to one of the amino acid residue types. In this study, we undertake a similar task using not only chemical shifts, but also TCs.
Previously, we used chemical shifts of IDPs deposited in BMRB (Romero et al. 2022) for the training step. However, such an approach would not be feasible for TCs since a relevant database does not exist. A possible solution is to use partial resonance assignment of the studied protein to train the algorithm, which will be later used to classify the not-assigned parts. There are many practical situations in which such a partial assignment is available. One of them is when we want to transfer the resonance assignment performed on a different sample of the same protein (e.g., measured under slightly different conditions). In such a case, some peaks are often well-separated and can be unambiguously assigned, while others are in crowded regions or severely shifted with respect to the original peak list. Another situation is that in the course of sequential assignment, some chains can be easily mapped on the protein sequence, but others are not sufficiently long or characteristic. Mapping the latter chains requires sophisticated methods of residue-type recognition, like LDA, that can also exploit the “easy part” of the data for training. Of course, an approach exploiting data from the same protein for training is justified only for relatively large proteins—for others, the number of assigned residues of each amino acid type can be too small to train the algorithm effectively. Also, the chemical shift values have to be dependent predominantly on the residue type, thus the approach applies to proteins of disordered nature. We demonstrate the method using spin systems from a Tau protein fragment of 239 residues containing 28 glycines and 26 prolines. Peaks corresponding to 141 residues from the peak list previously deposited in BMRB (entry 28065) fitted well to our spectra acquired at \(5^\circ\)C and were used for training. As discussed below, the ambiguities in assigning the 19 spin systems have been solved using LDA. For training and testing, we considered only residues for which complete sets of chemical shifts (\(}^}}\), N, C\(^\), \(}_\), \(}_\)) could be clearly found from our spectra. The glycines were excluded from the analysis since their assignment was, as usual, rather obvious. The remaining 22 resonances (not counting the N-terminal one) were missing.
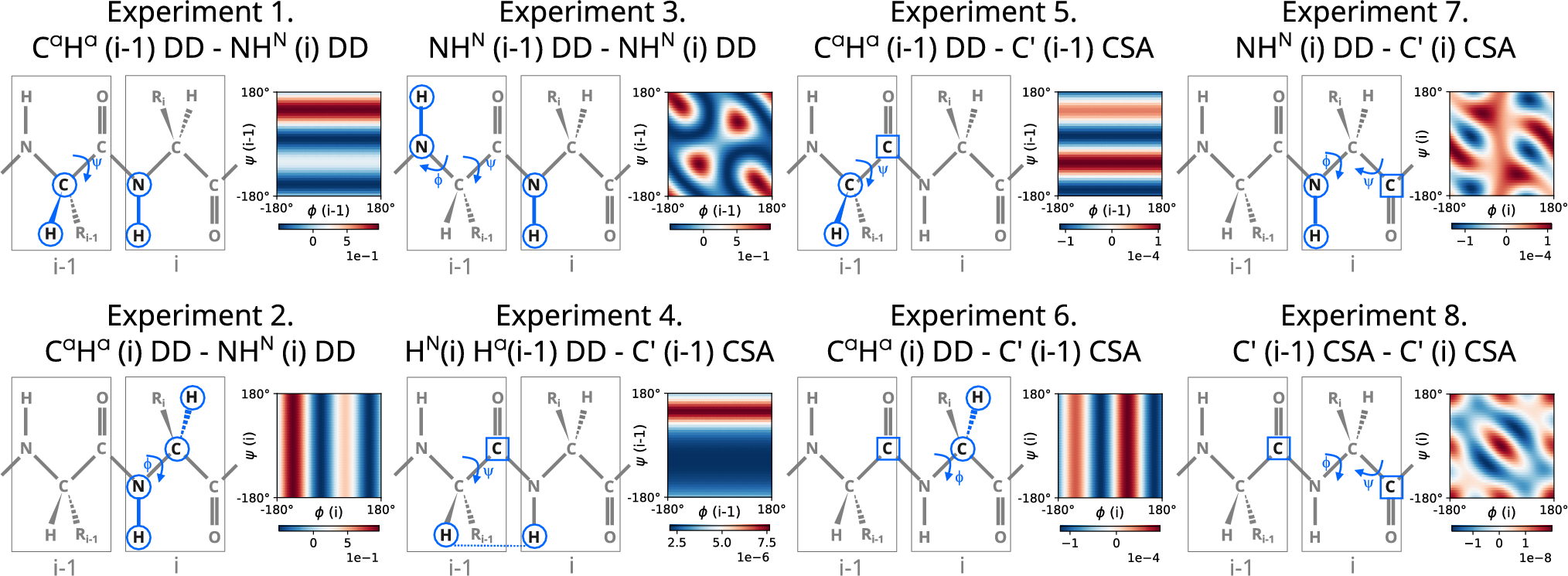
Figure 2 shows the results of using LDA on a 1-239 Tau fragment with and without TCs. We constructed six sets of CSs and TCs of different nuclei: subset (i) \(}^}}\), N, C\(^\), \(}_\) CSs; subset (ii): \(}^}}\), N, C\(^\), \(}_\) CSs and \(\hbox _\); subset (iii): \(}^}}\), N, C\(^\), \(}_\) CSs and \(\hbox _\) and \(\hbox _\); subset (iv): \(}^}}\), N, C\(^\), \(}_\) and \(}_\) CSs; subset (v): \(}^}}\), N, C\(^\), \(}_\), \(}_\) CSs and \(\hbox _\); subset (vi): \(}^}}\), N, C\(^\), \(}_\), \(}_\) CSs and \(\hbox _\) and \(\hbox _\).
Adding \(\hbox _\) to a set of \(}^}}\), N, C\(^\), and \(}_\) CSs (Fig. 2B) allows unambiguously recognizing lysine, leucine, and glutamine residues, which were not recognized by CSs only. Although, in one case, adding \(\hbox _\) causes misclassification of isoleucine residue (I151 is recognized as valine), the problem is solved by adding the \(\hbox _\). We get better results in subset (iii) than in subset (iv). Thus, when \(}_\) CS is not available, good variable-temperature data (e.g., for three different temperatures) can replace it. The most efficient is a subset (vi) (Fig. 2F). Generally, it correctly classifies amino acid residues, except for arginine, which is assigned to three classes. Nonetheless, the arginine has the highest probability (above 50%) of these three. As can be seen, the subset (vi) is only slightly better than (v) but requires collecting more data at higher temperatures, which is time-consuming and may be problematic in the case of not stable protein. Thus, we will use subset (v) for further examples discussed below. It is not crucial at which temperatures the spectra are acquired as long as differences in chemical shifts are residue type-specific, the protein is stable and amide proton chemical exchange does not hamper the measurement.
Notably, even when only CSs are used, training based on different parts of the Tau protein is optimal and increases classification efficiency compared to BMRB-based training discussed in our previous work (Romero et al. 2022) (see Supplementary Information Fig. S1). This might be caused by the temperature used in our experiment (\(5^\circ\)C) being very different from the typical temperatures in the BMRB entries from the training set.
Fig. 2
Results of linear discriminant analysis of CSs and TCs of 19 Tau 1-239 residues. The residues for which peak list transfer (from BMRB entry 28065) was ambiguous are presented. A subset (i): \(}^}}\), N, C', \(}_\); B subset (ii): \(}^}}\), N, C', \(}_\) and their \(\hbox _\); C subset (iii): \(}^}}\), N, C', \(}_\) and their \(\hbox _\) and \(\hbox _\); D subset (iv): \(}^}}\), N, C', \(}_\) and \(}_\); E subset (v): \(}^}}\), N, C', \(}_\), \(}_\) and their \(\hbox _\); F subset (vi): \(}^}}\), N, C', \(}_\), \(}_\), and their \(\hbox _\) and \(\hbox _\)
Another example application of LDA, besides peak list transfer, is mapping spin-system chains formed during sequential assignment on the protein sequence. The process is generally easier for long chains containing residues with characteristic chemical shifts (i.e., alanines, glycines, serines, and threonines). However, the chains are often interrupted when peaks are missing due to fast nuclear relaxation, chemical exchange, peak overlap, or lack of \(}^}}\) at proline residues. Unambiguous mapping of such short chains in a large protein is often difficult.
In the studied Tau fragment, several short chains between prolines were present. Figure 3 compares the efficacy of amino acid type recognition in these chains using LDA with three different kinds of training data: CSs from BMRB, and CSs from the same protein (Tau 1-239) with and without \(\hbox _\). We used the same training data for the latter two as for Fig. 2. Some of the short chains could not be mapped using LDA with CS-only BMRB-based training (Fig. 3, left side). In contrast, by training using data from the same protein in three of the shown cases (Fig. 3, panels C, D, E) the chains could be correctly mapped. For chains shown in Fig. 3a, b, the ambiguity still remains but is resolved by \(\hbox _\).
Let us discuss the short-chain identification from Fig. 3 in more detail. For the chain shown in panel A), LDA trained with chemical shift data from BMRB wrongly classifies the 211Arg, although a complete set of chemical shifts for this residue is available (\(}^}}\), N, C', \(}_\) and \(}_\)). The correct classification has the second highest probability (26%). Using training data from the same protein increases it by 10%, but still, the classification is wrong. The additional use of \(\hbox _\) resulted in the correct amino acid recognition (at the level of 57%). Another residue in the same chain—212Thr—is also misclassified if CS-only data is used (although only \(}^}}\), N and \(}_\)), but with \(\hbox _\), the 100% correct classification is achieved). A similar scenario is repeated for the 215Leu from Fig. 3b). Examples shown in Fig. 3c–e) present the superiority of the “same protein” approach over BMRB-based training. For all residues, the correct classification is better with the former approach. 217Thr, 50Thr, and 181Thr are properly classified only using LDA trained on chemical shifts from the same protein. Importantly, these are the spin systems with incomplete sets of chemical shifts (only \(}^}}\), N and \(}_\)). The additional use of \(\hbox _\) improves the correct classification even more.
Fig. 3
Comparison of LDA-based amino acid type recognition in short chains using 3 training data sets: BMRB, chemical shifts from the same protein (Tau 1-239) with and without TCs. LDA was performed using subset (iv): \(}^}}\), N, C', \(}_\), \(}_\); and subset (v): \(}^}}\), N, C', \(}_\), \(}_\), and their TCs. The recognitions with probability scores exceeding 10% are shown, and the correct residue type is marked in bold. Panels A–E show the recognition of different short chains
Comments (0)