
Remember me
The WHO fifth edition of Soft Tissue and Bone Tumours was published in 2020.1 Since then, excellent review papers have aimed to summarise emerging entities, some harbouring associated fusions. Fusion-associated entities recently summarised in a facetiously titled review by Folpe included: KMT2A-rearranged sarcoma, PRRX::NCOAx fibroblastic tumours, EWSR1::PATZ1 sarcomas, BRAF-altered infantile fibrosarcoma-like lesions, NUTM1-rearranged colorectal sarcomas, FN1-rearranged chondroid lesions and giant cell tumours of soft tissue with HMGA2::NCOR2 fusions.12 Emerging mesenchymal cutaneous neoplasms with gene fusions were recently summarised by Fischer and Papke, which included: nested glomoid neoplasm with GLI1 alterations, clear cell tumour with melanocytic differentiation and ACTIN::MITF translocation, melanocytic tumour with CRTC1::TRIM11 fusion, PLAG1-rearranged fibroblastic tumour and superficial ALK-rearranged myxoid spindle cell neoplasm.13 More recently, Towery and Papke have reviewed the clinicopathological features of recently described tumours identified on morphology and compared them to recently described entities based on next-generation sequencing, such as EWSR1::PATZ1 and GLI1-altered mesenchymal neoplasms.14
To further update additional emerging neoplasms, this review will include: clear cell stromal tumour of the lung (CCSTL) with YAP1::TFE3 fusion, GAB1::ABL1 spindle cell neoplasms, NUTM1-rearranged sarcomas, NR1D1-rearranged sarcomas and calcified chondroid mesenchymal neoplasms (CCMN). These emerging entities have been summarised in a morphological based table (online supplemental table 2) with histological mimics and respective PubMed IDs.
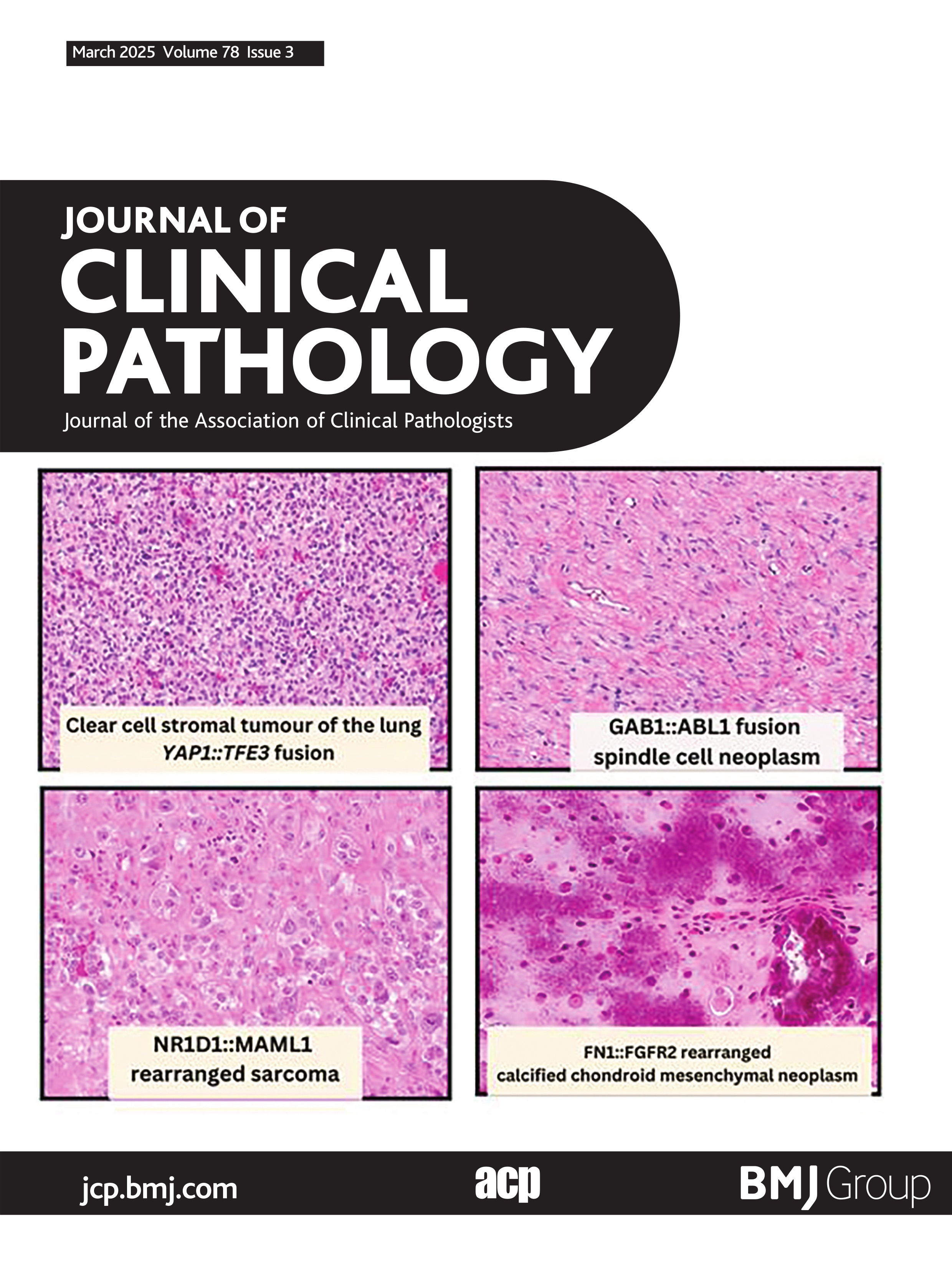
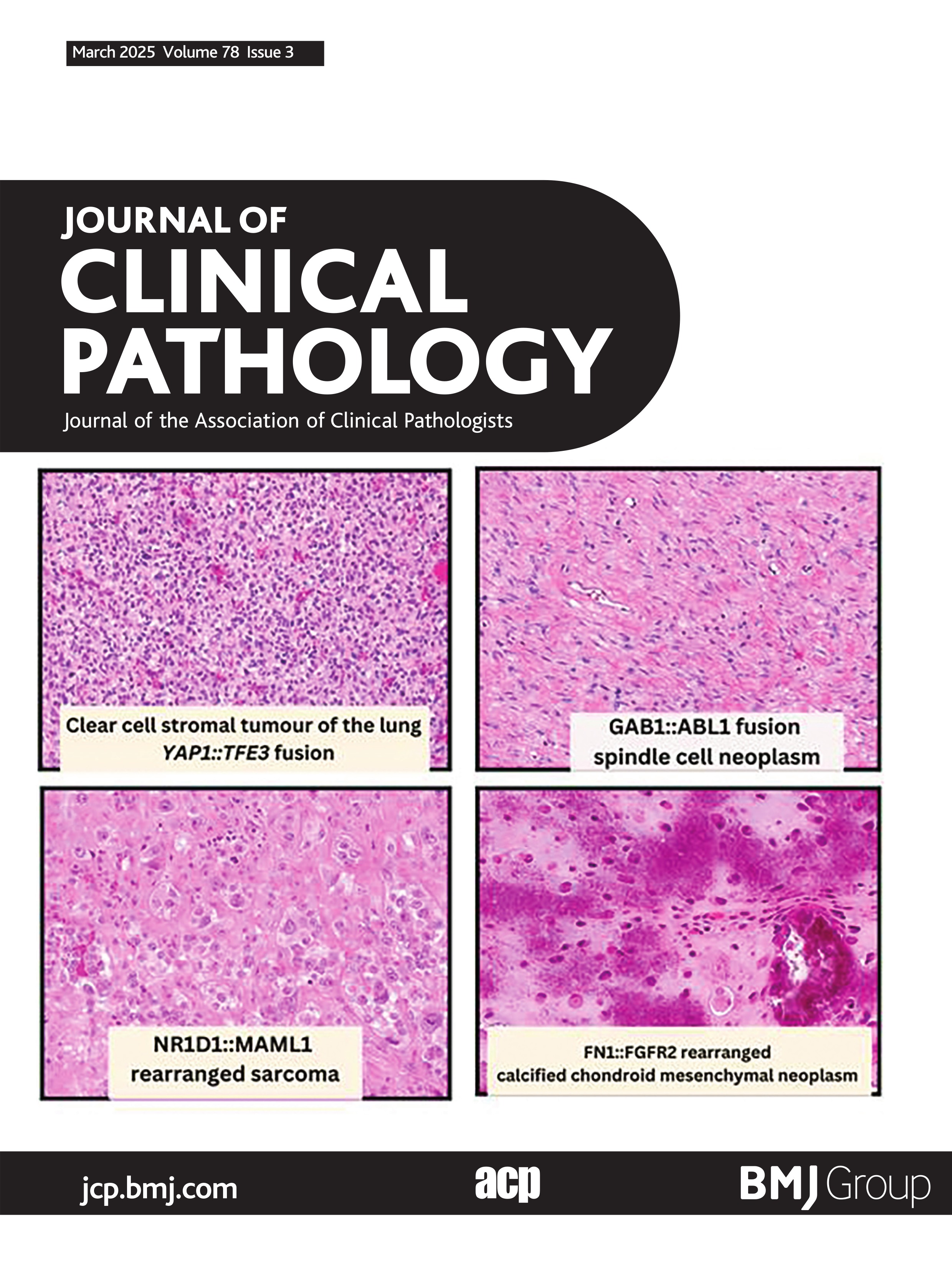
CCSTL with YAP1::TFE3 fusionCCSTL was first described in 2013 by Falconieri et al.15 Due to morphological overlap to haemangioblastoma, the two cases were coined ‘Hemangioblastoma-like clear cell stromal tumor of the lung’,15 although the tumour cells lacked the classic immunophenotype of central/peripheral haemangioblastoma: NSE, inhibin and S100.16–18 Subsequently, in 2020 Lindholm and Moran published a series of five cases with similar morphological findings.19 In 2021, Agaimy et al identified YAP1::TFE3 fusions in four of five cases of CCSTL.20 Since then, other groups have published YAP1::TFE3 fusions in CCSTL,21–23 totalling 10 molecularly confirmed CCSTL with YAP1::TFE3 fusions reported in the literature. Morphological features of these neoplasms include a vague nested to sheet-like architecture of uniform medium-sized epithelioid to spindle cells with clear cytoplasm and small monomorphic nuclei within a variably hypervascular stroma (figure 1). The cytoplasmic clearing is described to have a more flocculent and granular quality than seen in other clear cell neoplasms.20 These tumours lack any specific line of differentiation and are all reportedly diffusely positive for TFE3 immunohistochemical stain.20–23 Scattered atypical cells have been seen in four cases in the form of hyperchromatic nuclei with marked pleomorphism.20 22 These CCSTL with YAP1::TFE3 fusions have all occurred in adults to date, with age ranging from 24 to 77 years old, with a slight female predominance (seven females, three males). Nine of eleven patients have clinical follow-up data available.21 Persistent disease over 4 years was noted in one patient. One of the nine patients had biopsy-proven hepatic and renal metastases. One patient died of disease at a 7-month follow-up.23 The remaining patients showed no evidence of disease, with varied lengths of clinical follow-up time.21–24
 Figure 1
Figure 1 Clear cell stromal tumour of the lung with YAP1::TFE3 fusion. (A) Low-power image showing well-circumscribed tumour within lung parenchyma (40×). (B) High-power image showing haphazardly arranged spindle cell neoplasm with eosinophilic to vacuolated/clear cytoplasm and surrounding vasculature (200×). (C) Strong nuclear positivity for TFE3 IHC (100×). (D) Schematic showing fusion of YAP1 exon 4 to TFE3 exon 7. Figure 1A–C courtesy of Dr Elizabeth Demicco (Toronto, Canada).
When confronted with clear cell neoplasms in the lung, primary pulmonary carcinomas with clear cell morphology will be first in consideration, subsequently with metastatic clear cell renal cell carcinoma, among others. PEComas often impart a clear cell morphology and a subset is TFE3-rearranged24 25; however, the myomelanocytic differentiation which is a hallmark of this entity is absent in CCSTL. Of note, this same fusion is seen in a subset of epithelioid haemangioendothelioma, although these tumours have a unique morphology of prominent eosinophilic cells and vasoformation, which is further supported by endothelial differentiation by immunohistochemistry.26 27
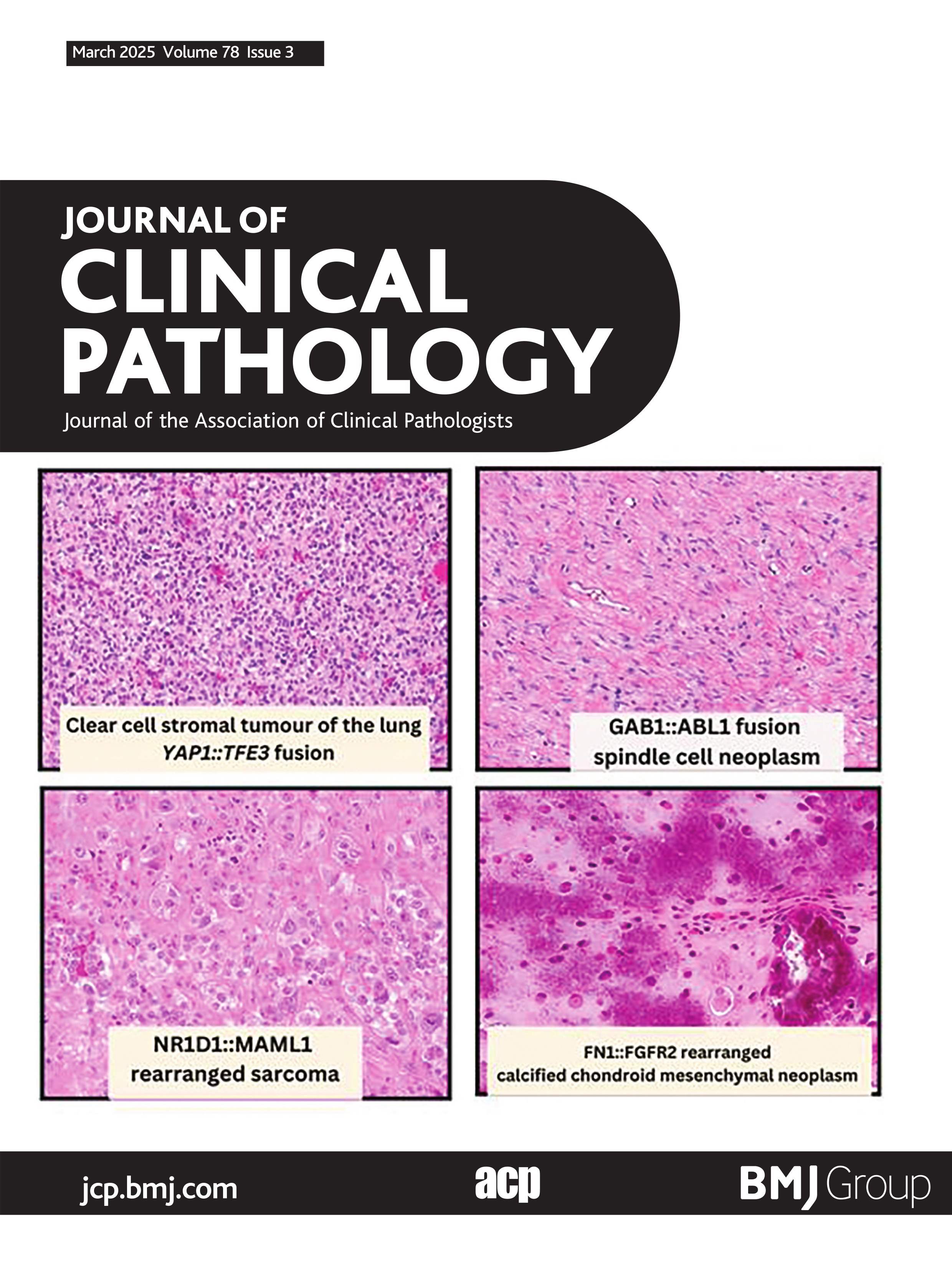
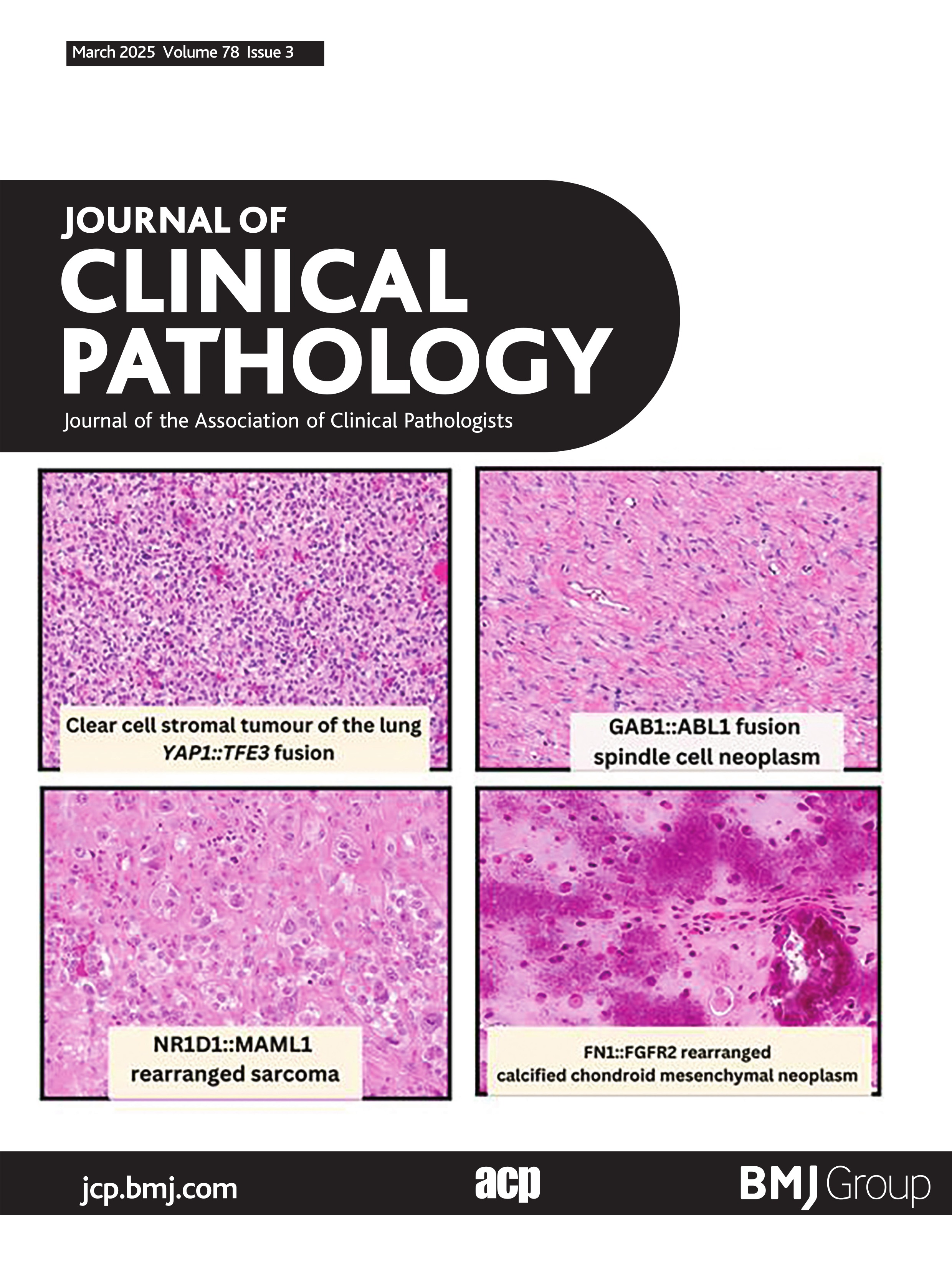
GAB1::ABL1 fusion spindle cell neoplasmVery recently Agaimy et al published an additional eight cases of soft tissue neoplasms harbouring GAB1::ABL1 fusions,28 adding to the previously five reported cases in the literature.29–32 These neoplasms were variably classified as soft tissue perineurioma, soft tissue angiofibroma, fibroblastic tumour not otherwise specified and solitary fibrous tumour/NTRK-like fibroblastic tumour. Histologically, these are poorly circumscribed hypocellular tumours which can entrap adipose tissue, fascia and skeletal muscle peripherally. The cells themselves are bland spindle to ovoid cells embedded in a fibrous to fibromyxoid stroma (figure 2). The vasculature is composed of elongated to focally staghorn-like thin-walled blood vessels. Vague whorling and storiform pattern, akin to perineurioma was noted in most cases. Generally, these are mitotically low neoplasms with no atypical forms, no cytological atypia, necrosis or lymphovascular invasion.28 These neoplasms showed variable expression of perineurial cell markers EMA (6/12), claudin1 (3/4), GLUT1 (4/7) and CD34 (8/11), but were less frequently S100 positive (3/12).28 Other lineage markers such as desmin, SMA, keratins and SOX10 were negative.28 Given the histological overlap with low-grade fibromyxoid sarcoma and variably solitary fibrous tumour it is important to note MUC4 and STAT6 immunohistochemical stains were negative in cases tested.28 Of note, pan-TRK showed weak expression in one of the three cases tested.28 The patient age of this group of neoplasms is of importance as 46% occur in the paediatric population (<18 years old) at the time of diagnosis, but with a wide age range, overall, from 9 to 76 years (median: 23 years).28 These are likely benign neoplasms given the lack of documented metastatic disease. Of the eight cases with clinical follow-up, none have shown metastatic potential; only one developed rapid recurrence within 1 month of excision and was subsequently re-excised.28
 Figure 2
Figure 2 GAB1::ABL1 fusion spindle cell neoplasm. (A) Low-power image showing tumour cells within a fibrous to edematous stroma (40×). (B) High-power image showing irregular fascicular growth of small bland-appearing spindle cells embedded within fibrous stroma (400×). (C) Claudin-1 IHC with positivity along perineurioma-like cytoplasmic processes (400×). (D) Electron microscopy of GAB1::ABL1 fusion spindle cell neoplasm showing long thin cytoplasmic processes. Inset bottom-left: prominent pinocytic vesicles. Inset top-right: cytoplasmic processes are joined by cell junctions. (E) Schematic showing fusion of GAB1 exon 6 to ABL1 exon 2.
Agaimy et al nicely summarised the lack of specificity in perineurial cell immunohistochemical stains, in particular claudin-1, arguing GAB1::ABL1 fusion spindle cell neoplasms represent a unique entity, although further studies are required. In our experience, ultrastructure of GAB1::ABL1 fusion spindle cell neoplasms do have perineurial differentiation, and we favour classifying these tumours as perineurioma with GAB1::ABL1 fusion (figure 2D). Low-grade fibromyxoid sarcoma and solitary fibrous tumour would be prudent to exclude when faced with the above cytomorphological features, which thankfully have helpful immunohistochemical support with MUC4 and STAT6, respectively. A more challenging differential diagnosis to consider is NTRK-rearranged spindle cell neoplasm, given the CD34/S100 coexpression that both these neoplasms share. However, the cellularity in NTRK-rearranged spindle cell neoplasms is often more pronounced, to the degree of hypercellularity seen in high-grade malignant peripheral nerve sheath tumour and infantile fibrosarcoma. Additional helpful histological features seen in NTRK-rearranged spindle cell neoplasms include prominent stromal collagen and keloid-like hyalinised perivascular rings, which have not yet been observed in GAB1::ABL1 fusion spindle cell neoplasms.
NUTM1-rearranged sarcomasNUTM1 rearrangements were initially associated with poorly differentiated squamous cell carcinomas in children and young adults, with a predilection for midline sites.33 34 NUTM1 rearranged neoplasms have now expanded beyond epithelial malignancy, and have been described in some haematolymphoid neoplasms, embryonal tumours of the central nervous system, adnexal tumours and undifferentiated sarcomas.35–38 CIC::NUTM1 rearranged sarcomas are excluded from this discussion, as these have been shown to cluster with CIC::DUX4 sarcomas showing small round blue cell morphology.39 40 Although there were some early reports of sarcomas harbouring BRD3/4::NUTM1 fusions, in general NUTM1-rearranged sarcomas harbour gene fusions with partners CIC and MAD family of genes (MGA, MXD1, MXD4 and MXI1).35 41–51 Other fusion partners reported in single cases include BCORL1 and ZNF532. 43 52 Interestingly NUTM1::MXD4 rearranged sarcomas have most commonly been reported in the colon (6/8 cases), with two cases being reported in the ovary and penile shaft.35 41 46 47 Awareness of the fusion partner in the context of a NUT-rearranged sarcoma may have future therapeutic implications, as non-BRD NUTM1-rearranged sarcomas may not respond to bromodomain and extra-terminal domain inhibitor therapy.53 54
These sarcomas span a wide age range (3–78 years of age) and anatomical sites (including viscera, colon, lung/chest wall, soft tissue of extremities, and single reports in the brain and bone). MAX-family rearranged sarcomas show a predilection for the gastrointestinal tract and lung.47 Histologically, as with other fusion-associated sarcomas, the neoplastic cells are monomorphic, varying from spindled, epithelioid and less frequently rhabdoid with abundant hyalinisation/amianthoid-like fibres (figure 3). The fibrosarcomatous and hyalinised/nested growth patterns are most commonly described in the MAX family-rearranged sarcomas.47 Immunophenotype is non-specific; however, NUT protein expression of variable intensity is a consistent feature.47 NUTM1-rearranged sarcomas have shown aggressive biological behaviour, with frequent metastases (only 7/27 (26%) of patients reported in the literature are alive without disease).
 Figure 3
Figure 3 MGA::NUTM1 rearranged lung sarcoma. (A) Low-power image showing cellular population of spindle cells with bands of hyalinised fibrosis (100×). (B) High-power photomicrograph shows short fascicles of spindle cells with scant cytoplasm, open vesicular chromatin and small nucleoli (400×). (C) Tumour cells show nuclear positivity for NUT IHC (200×). (D) Schematic showing fusion of MGA exon 22 to NUTM1 exon 2.
The differential diagnosis of this neoplasm varies based on anatomic site and age. Hypercellular monomorphic spindle cell neoplasms in the differential diagnosis would include solitary fibrous tumour, synovial sarcoma, malignant peripheral nerve sheath tumour and gastrointestinal stromal tumour. In the presence of epithelioid morphology and abundant hyalinised background, sclerosing epithelioid fibrosarcoma would be an important diagnosis to exclude. Prominent epithelioid and rhabdoid morphology includes spectrum of SWI/SNF deficient neoplasms. Sarcomatoid carcinomas and mesotheliomas should also be considered in the presence of variable keratin expression. Judicious use of immunohistochemical stains will help in excluding the above differential diagnoses while ancillary molecular testing results are pending.
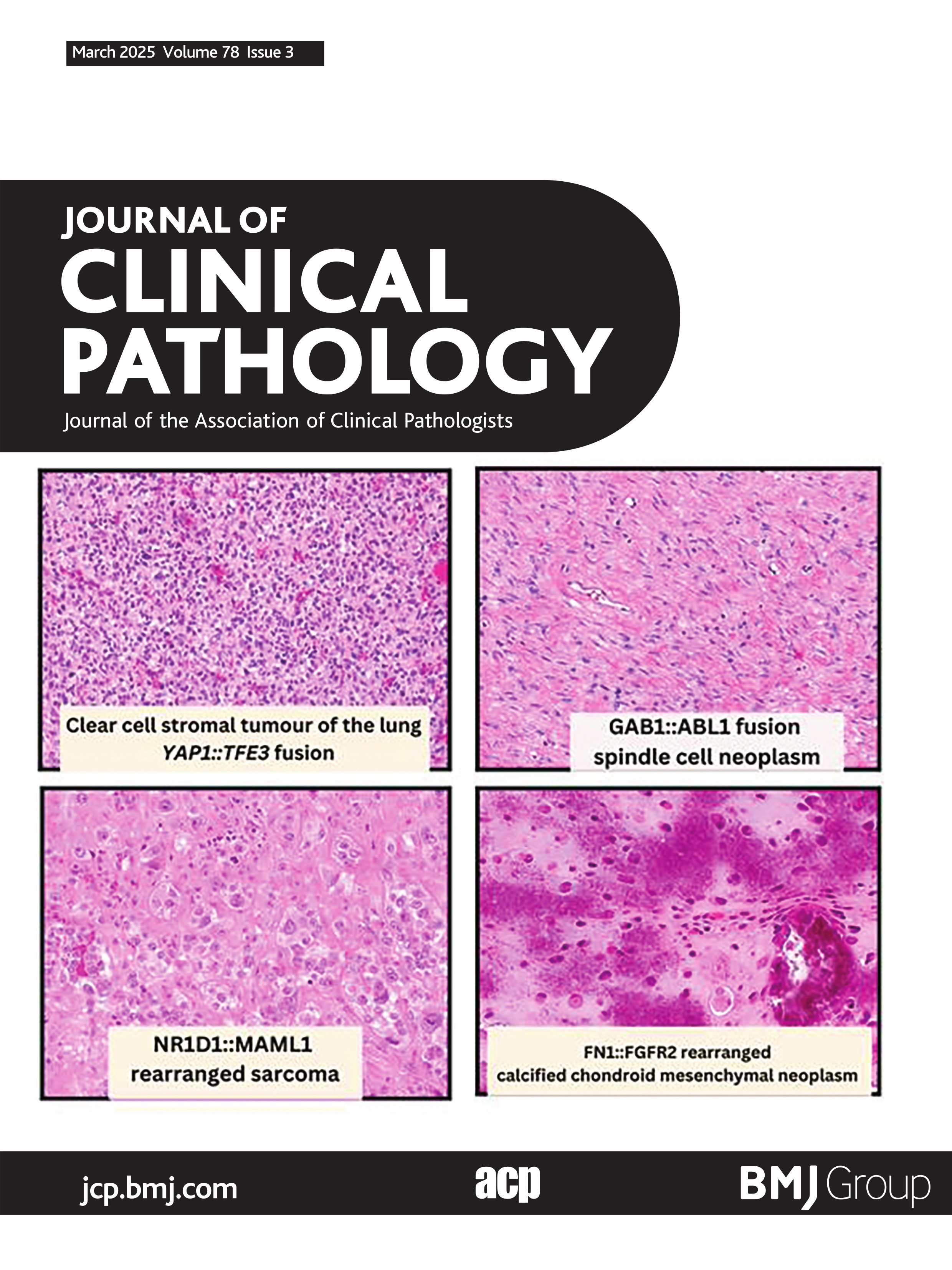
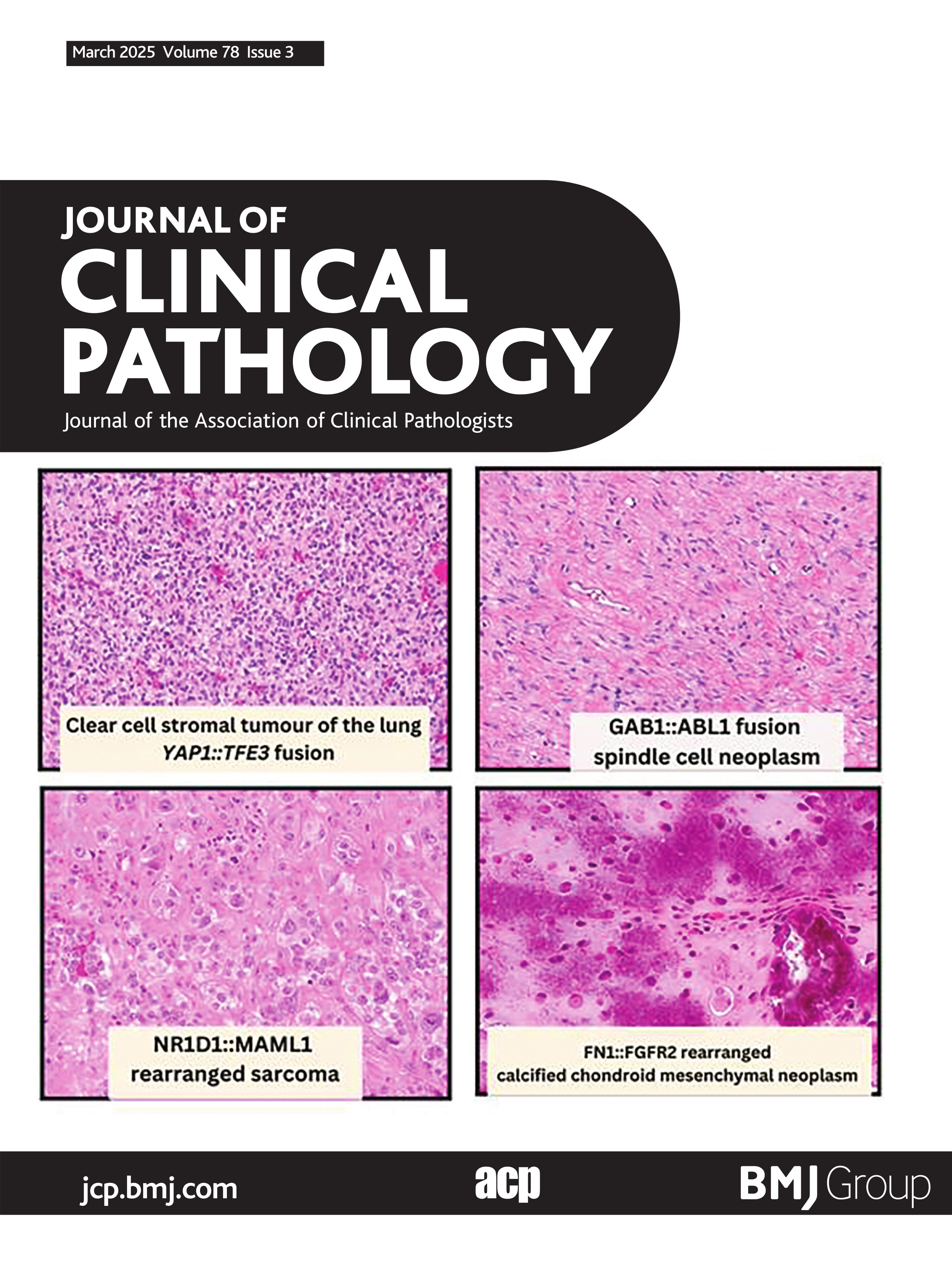
NR1D1-rearranged sarcomasKomatsu et al published the first NR1D1-rearranged sarcoma of soft tissue in a 10-year-old boy.55 Currently seven NR1D1-rearranged sarcomas have been described in the literature.55–58 The largest case series to date was by Lacambra et al, which described four cases all of which occurred in females.57 This novel fusion identified in these neoplasms has included NR1D1 as a fusion partner with MAML1 (5/7 cases) and MAML2 (2/7 cases). Anatomically these tumours frequently involve the soft tissue of limbs (5/7), foot (1/7) and abdominal wall (1/7). With regards to soft tissue compartments, tumours frequently involve the subcutaneous compartment with two cases showing deep soft tissue involvement and one with superficial bone involvement. The age range varies from 10 to 70 years old (median 62 years), with a near equal male to female ratio (three male:four female). For the six patients with available follow-up data, half showed pulmonary metastases. Two patients died of disease (2/6)—one patient died 3 years after surgery and several chemotherapies for carcinoma of unknown primary after multiple metastases to the lungs and mediastinal lymph nodes, the other patient died of disease 13 months after diagnosis after palliative radiation to an unresectable tumour.57 One patient is presumed alive with disease (1/6) and half of documented patients have shown no evidence of disease (3/6).
Morphologically these tumours show a predominantly epithelioid morphology, with a minor spindle cell component (figure 4). Architecturally these tumours can show pseudopapillary, nested and sheet like architecture. Corded growth has also been documented.57 Three cases have shown scattered cells with intracytoplasmic vacuoles.57 58 Warmke et al recently summarised the immunophenotypic findings of this sarcoma.58 Keratin expression has been seen in all seven reported cases, with three cases showing diffuse staining and four cases showing focal staining (figure 4C). One case showed rare focal expression of S100 and one case showed both FOSB and ERG positivity, raising the possibility of pseudomyogenic haemangioendothelioma.58 Other reported markers (CK20, CK5/6, CD34, CD31, desmin, SMA, SOX10) were negative. Besides pseudomyogenic haemangioendothelioma, the differential diagnosis also includes alveolar soft part sarcoma, epithelioid sarcoma, PEComa, myoepithelial tumours and poorly differentiated carcinomas, which can be distinguished based on judicious use of immunohistochemical stains and identification of the underlying NR1D1 fusion event.
 Figure 4
Figure 4 NR1D1::MAML1 rearranged sarcoma. (A) Low-power image showing multinodular tumour cells separated by fibrous tissue and thin fibrous pseudocapsule (100×). (B) High-power photomicrograph showing plump epithelioid cells with prominent nucleoli, abundant vacuolated cytoplasm and scattered neutrophils embedded in fibrous stroma (400×). (C) Tumour cells show multifocal keratin expression (AE1:AE3, 400×). (D) Schematic showing fusion of NR1D1 exon 6 to MAML1 exon 2. Figure 4A–C courtesy of Dr Karen Fritchie (Cleveland, OH).
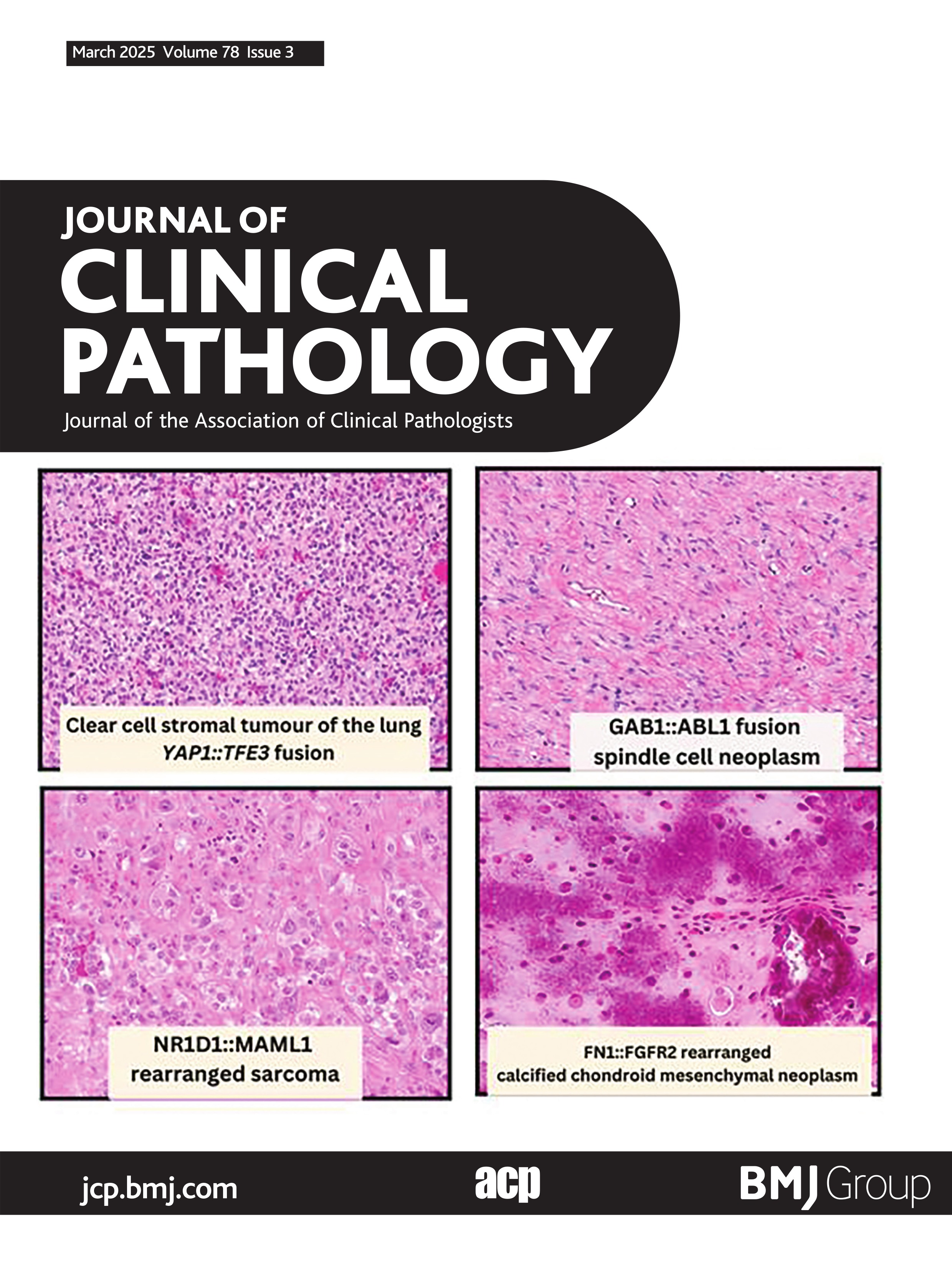
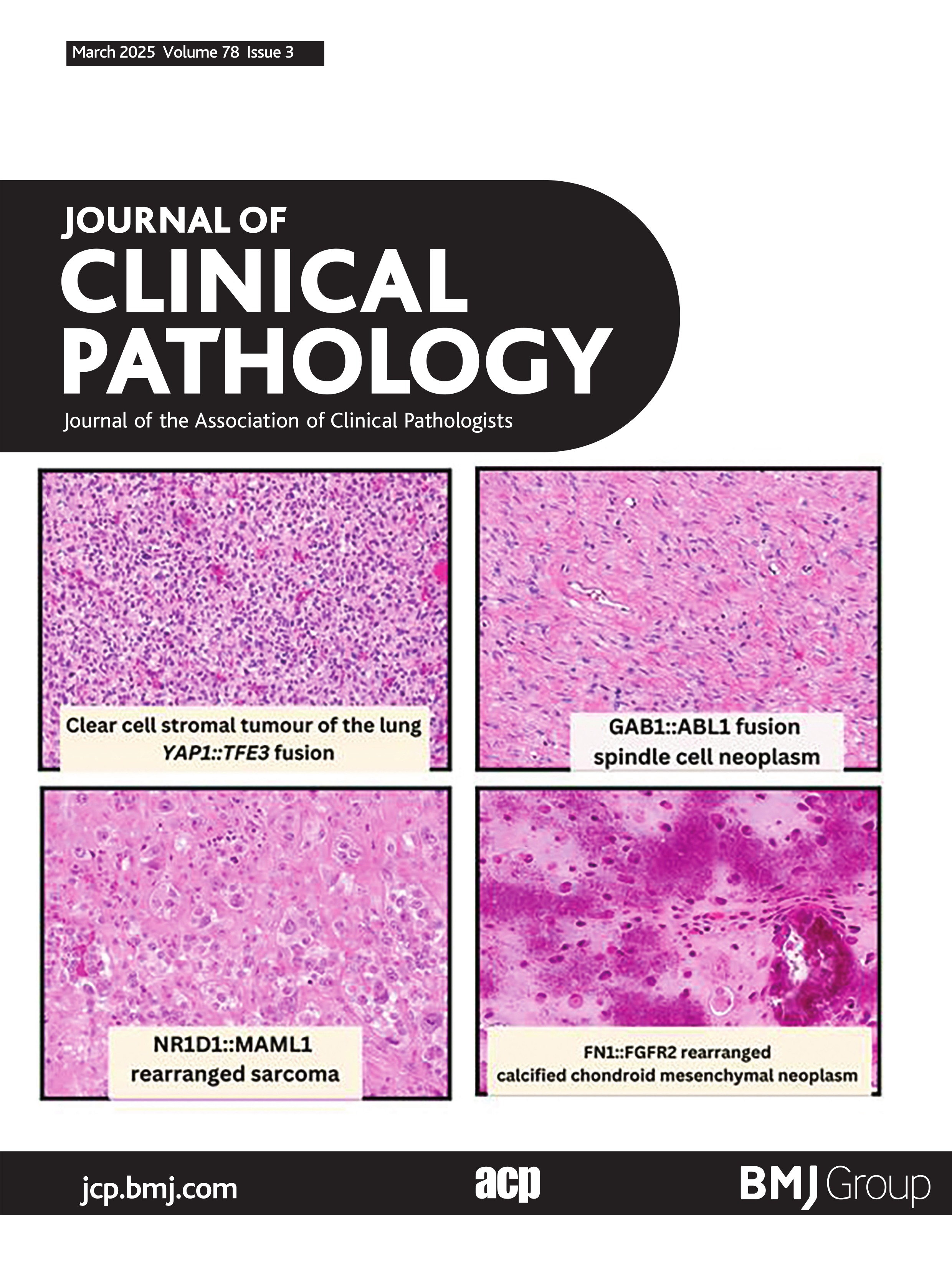
Calcified chondroid mesenchymal neoplasmsSince the first description by Liu et al of this group of neoplasms termed ‘calcified chondroid mesenchymal neoplasms (CCMN)’, there has subsequently been a total of 87 described in the literature.59–66 This group of neoplasms is histologically characterised by lobular growth of neoplastic cells with intervening fibrous septae (figure 5). The neoplastic cells show polygonal-epithelioid and occasionally spindled morphology, with amphophilic cytoplasm with mild cytological atypia. These tumour cells are embedded in an abundant fibromyxoid to chondrohyaline matrix, with characteristic calcification patterns, described as ‘chondroblastoma-like’, ‘grungy’ and ‘lace-like’. The immunophenotype is variable and non-specific (refer to online supplemental table 2). Additional histological features which are variably present includes multinucleated giant cells and calcium pyrophosphate deposition. All but two cases have been described in adults; the paediatric cases occurred in an 11-year-old and a 14-year-old.60 66 These tumours have a predilection for the distal extremities (63/87) and temporomandibular joint (17/87). Other documented locations include the parotid gland,60 right hamstring tendon,62 right gastrocnemius,64 external auditory canal59 and the hip.65 66 These tumours behave in a benign fashion, with no documented metastases and two cases with available clinical data showing local recurrence60 and four cases showing local aggressiveness.66 Recurrent fusion events have been documented in 51 of 87 cases. Fusion events have included FN1 (40/51), PDGFRA::USP8 (9/51) and one case of FGFR1::PLAG1 (1/51)59 and one case of COL1A2::MIR29B1 (1/51).66 Additional cases of CCMN were recently reported by Benard et al which underwent extensive molecular analyses.66 Interestingly, a subset of temporomandibular joint CCMN’s showed tenosynovial giant cell tumour-like features showed FN1::TEK fusions.66 The subset of PDGFRA::USP8 driven CCMN’s show a predilection for larger joints and show multiple foci of bone formation, these features being exhibited in half of the cases.64–66 Other previously described entities within the histomorphological spectrum include the chondroblastoma-like variant of soft tissue chondroma and chondroid tenosynovial giant cell tumours, in which there is currently debate as to whether these two entities are best classified under the umbrella term of calcified chondroid mesenchymal neoplasms.
 Figure 5
Figure 5 FN1::FGFR2 rearranged calcified chondroid mesenchymal neoplasm. (A) Low-power photomicrograph showing abundant calcification and surrounding chondromyxoid stroma (100×). (B) High-power image showing grungy calcifications, calcium pyrophosphate crystals and plump epithelioid to plasmacytoid cells within chondromyxoid stroma (400×). (C) Abundant variably polarising rhomboid to elongated crystalloids morphologically consistent with calcium pyrophosphate (400×). (D) Schematic showing fusion of FN1 exon 39 to FGFR2 exon 3.
Comments (0)