
Remember me



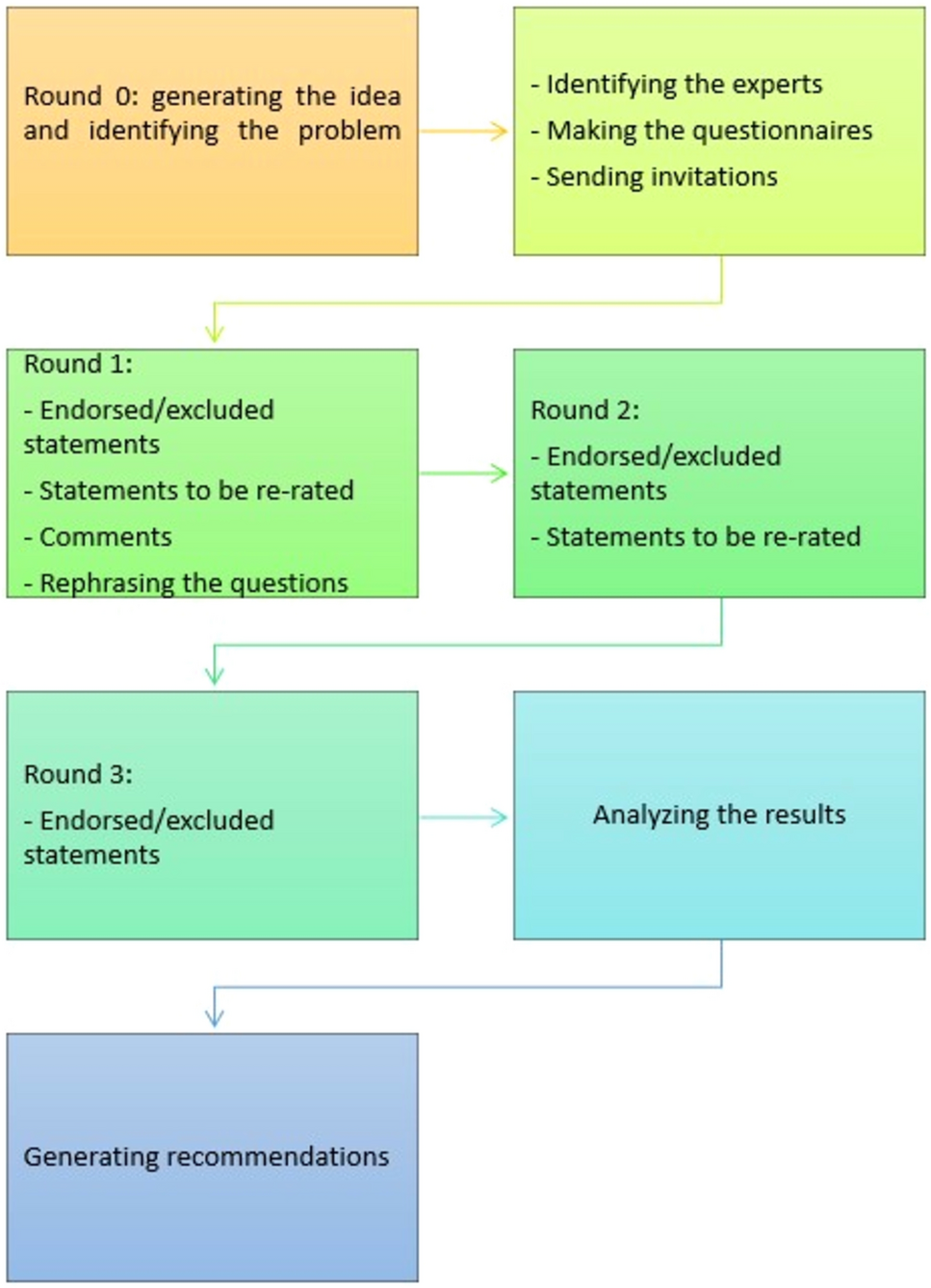
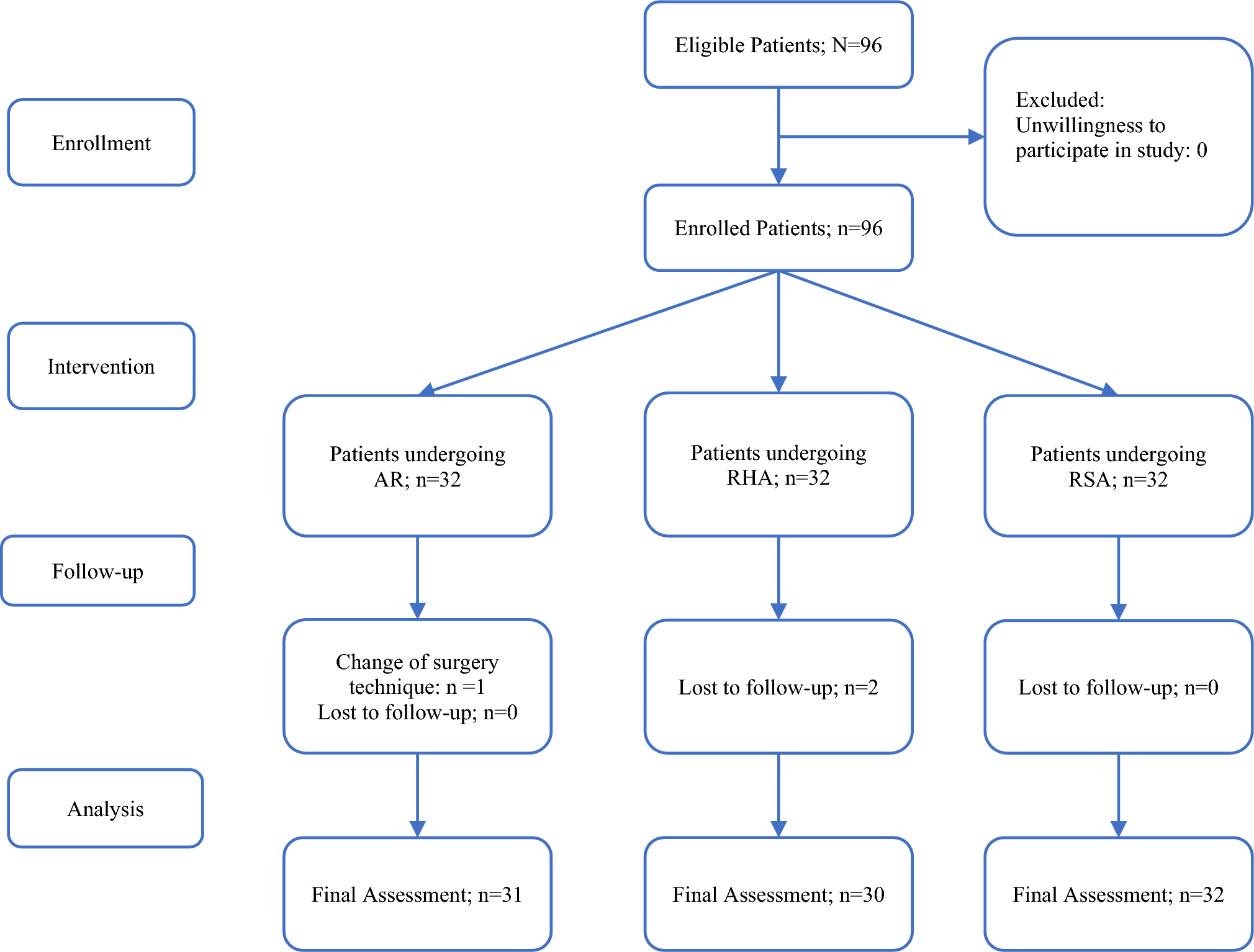
This is a prospective, open-label, multicenter, non-inferiority RCT of two parallel groups, conducted at the Shizuoka Cancer Center and 31 other institutions in Japan. A flowchart of the BANDIT trial is displayed in Fig. 1. The trial protocol was designed according to the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines [35]. The SPIRIT flow diagram is shown in Fig. 2.
Fig. 1
Flowchart of the study design. ESD, endoscopic submucosal dissection; ESMR-L, endoscopic submucosal resection with a ligation device
Fig. 2
Schedule of enrollment, interventions, and assessments as per Standard Protocol Items: Recommendations for Interventional trials (SPIRIT). ESD, endoscopic submucosal dissection; ESMR-L, endoscopic submucosal resection with a ligation device
ApprovalsThe study protocol was approved by the Certified Review Board (CRB) of Shizuoka Cancer Center (CRB4180010; Nov 24, 2021). The trial was registered in the Japan Registry of Clinical Trials (jRCTs042210124; Jan 6, 2022).
Eligibility criteriaInclusion criteriaPatients should meet all of the following criteria.
1) Patients with a lesion endoscopically diagnosed as rectal NET ≤ 10 mm.
2) Patients who are at least 20 years old at the time of obtaining consent.
3) Written informed consent should be provided based on the free will of patients after they have thoroughly understood the instructions provided regarding study participation.
Exclusion criteriaPatients are excluded if they met any of the following criteria.
1) Patients whose clinical course cannot be followed up to 28 days after treatment.
2) Patients with a recurrent lesion.
3) Patients with inflammatory bowel disease or colorectal polyposis.
4) Patients with coagulation dysfunctions.
5) Patients with severe infectious disease.
6) Patients on dialysis.
7) Patients who cannot discontinue antithrombotic agents based on the guidelines for gastroenterological endoscopy in patients undergoing antithrombotic treatment [36, 37].
8) Pregnant patients.
9) Other cases determined to be unfit for the study by the investigator.
Informed consent procedurePatients are screened for eligibility by endoscopists at each institution based on the above-mentioned criteria. The patients will receive detailed information about the trial from a member of the research team at their respective institutions. Ample time will be provided to patients for considering participation in the trial. Written informed consent will be obtained from each patient before enrollment.
RandomizationUpon enrollment of eligible patients by the investigators, a randomization process will be conducted to assign patients to either the ESD or the ESMR-L group. This randomization will follow a 1:1 allocation ratio and will be performed using a web-based registration system available at Shizuoka Cancer Center, which is accessible 24 h a day. This study will use minimization method. Adjustment factors are participating institutions and endoscopist expertise levels (expert or non-expert).
BlindingOwing to the nature of the trial intervention, blinding endoscopists, and patients is not feasible. Endoscopists should be aware of the allocated group at the time of treatment, and patients may become aware of their assigned group during the course of treatment.
Trial interventionsEach patient with a rectal NET is treated using the assigned endoscopic procedure. Only one target lesion is treated per patient. Hospitalization is recommended, however, it is not mandatory.
Standard treatment group (ESD group)ESD is a procedure involving a mucosal incision and submucosal dissection using ESD knives [38]. ESD knives are specified to the following; DualKnife J (KD-655, Olympus Medical Systems Corp, Tokyo, Japan), FlushKnife BT-S (DK2620J, Fujifilm, Tokyo, Japan), HookKnife J (KD-625, Olympus Medical Systems Corp), and Jet B-knife (Zeon Medical Inc, Tokyo, Japan). The endoscope is selected at the discretion of the operator. CO2 insufflation is recommended. This trial does not specify the type and mode of the electrosurgical unit, the use of a distal attachment, or the type of solution for submucosal injection. Traction devices are allowed to use. In cases of intraprocedural bleeding, hemostasis is achieved through coagulation with the tip of the ESD knife or hemostatic forceps. If intraprocedural perforation or muscle injury occurs, the resection site will be clipped.
Trial treatment group (ESMR-L group)ESMR-L will be performed using an endoscope equipped with a band ligator device (Pneumoactivate EVL device, SB-Kawasumi Laboratories, Inc, Tokyo, Japan) [9]. The lesion will be removed using submucosal injection. Subsequently, the lesion will be aspirated into the ligator device, followed by the deployment of the elastic band. Afterward, snare resection will be performed below the band using a blended current. The endoscope is selected at the discretion of the operator. CO2 insufflation is recommended. The type of solution used for the submucosal injection and the type of snare employed are not specified. In cases of bleeding immediately after resection, hemostasis is achieved by coagulation with the tip of the snare, hemostatic forceps, or clipping. If intraprocedural perforation or muscle injury occurs, the resection site will be clipped.
Intervention success and failureTreatment is considered successful when the operator determines that the specimen has been retrieved without any residual lesions. In cases where the operator deems it impossible to complete the resection using the assigned treatment and specified devices, the treatment protocol will be terminated. Additional treatment options may be allowed after termination.
Restrictions after resectionIn the absence of perforation or muscle injury, prophylactic clipping or covering methods, such as polyglycolic acid sheets and fibrin glue, are not recommended, however, they are permitted. If the operator applies prophylactic clipping or covering methods, they are required to apply the same method to both the ESD and ESMR-L groups. Prophylactic endoscopic coagulation is permitted only for visible vessels.
Pathological assessmentResected specimens are immediately fixed on a panel using pins in a manner that aligns with the tumor diameter. To evaluate the maximum cut surface and vertical margins, the specimens are fixed in 10% buffered formalin and sectioned at the deepest part of the tumor. Resected specimen size, tumor size, classification (NET G1, G2, G3, neuroendocrine carcinoma [NEC]), invasion depth, lymphatic invasion, vascular invasion, horizontal margins, and vertical margins are recorded. Final pathological diagnoses will be determined by pathologists at each participating institution utilizing the WHO Classification of Tumors, 5th Edition (2019) and the Japanese Classification of Colorectal Carcinoma [39].
Additional colectomy with lymphadenectomy may be considered for the following; tumor size ≥ 1 cm or grade higher than G2, positive vertical margins, muscularis propria invasion, or lymphovascular invasion [40].
Outcome measuresThe primary endpoint (for non-inferiority)R0 resection rate is defined as the absence of tumor involvement in both the horizontal and vertical margins, as confirmed histologically in the resected specimen. Evaluation of the horizontal and vertical margins should be performed on the plane where the NET or NEC is present. Margins without NET or NEC, irrespective of the distance, are considered to be tumor-free. Subsequently, R0 resection will be assessed by the operator based on a pathological report.
Secondary endpointsEn bloc resection is defined as the complete removal of the lesion in a single piece without residual visible tumors.
Procedure time refers to the duration from submucosal injection to completion of the resection. It does not include the time required for prophylactic clipping, covering methods, or endoscopic coagulation of visible vessels after resection.
4. Adverse events (AEs)
Adverse events will be monitored and recorded, including the following:
a. Delayed bleeding: bleeding that occurs within 28 days after colonoscopy withdrawal that requires hemostasis.
b. Intraoperative perforation: defect in the muscular layer observed during the procedure, or the presence of free air in the peritoneal cavity on imaging studies performed within 12 h after treatment.
c. Delayed perforation: bowel perforation within 28 days after the procedure without intraoperative perforation, confirmed by the presence of free air in the peritoneal cavity on imaging studies performed > 12 h after treatment.
d. Electrocoagulation syndrome: localized abdominal tenderness and fever (≥ 37.6℃), or inflammatory response without definite evidence of perforation.
5. Number of hospitalization days
Outpatient treatment will be considered as 0 days.
The costs associated with the ESD knife, band ligator device, injection needle, distal attachment, snare, traction device, clips, solution for submucosal injection, and antibiotics are calculated.
Patients who undergo closure of the resection site using clips or detachable snares will be compared with those without closure.
The outcomes of expert endoscopists, defined as those with experience in > 40 colorectal ESD procedures [38, 41]. will be compared with those of non-expert endoscopists.
The potential factors contributing to the failure of achieving R0 resection, including treatment method, tumor location, tumor size, previous biopsy, and endoscopist expertise, will be analyzed.
Serious adverse eventsThe following AEs are classified as serious: AEs resulting in death or posing a life-threatening risk, AEs requiring rehospitalization for treatment or extension of the hospitalization period, AEs leading to persistent or marked disability, and AEs with the potential to cause birth defects in the offspring. AEs will be assessed and graded according to the Common Terminology Criteria for Adverse Events Version 5.0. If a serious AE occurs, the investigators should report it immediately to the principal investigator. If the principal investigator determines a causal relationship between a serious AE and the protocol treatment, a serious AE report will be submitted to the CRB and the hospital director. In addition, information regarding the occurrence of serious AEs will be shared promptly with all the investigators.
Sample size calculationThe sample size is calculated based on the primary outcome parameter, the R0 resection rate, to assess the non-inferiority of ESMR-L to ESD. These assumptions are based on previous studies and systematic reviews [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32], with an expected R0 resection rate of 95.2% in the ESD group and 95.3% in the ESMR-L group. The non-inferiority margin is determined following the FDA Non-Inferiority Clinical Trials to Establish Effectiveness Guidance for Industry 2016 (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/non-inferiority-clinical-trials). This FDA guidance illustrates the use of half the difference between the standard treatment and placebo as the non-inferiority margin. According to this guidance, the non-inferiority margin is set at 8% as half the superiority effect of the standard treatment (ESD, 95.2%) over the previous control arm (conventional EMR, the R0 resection rate 78% [29]). Based on these assumptions, 226 participants are required to assess the non-inferiority of the ESMR-L, with a one-sided significance level of 0.05 and a power of 80%. Considering a potential dropout rate of 15%, including patients without NET or NEC in the resected specimen and those who refused consent or were lost to follow-up, 266 patients will be included, with 133 patients in each arm. To achieve sufficient participant enrollment, 32 Japanese institutions will be included in this trial.
Statistical analysesThe main analysis will be conducted using the full analysis set (FAS). The FAS is defined as the population of enrolled participants, excluding any of the following criteria: (1) any violation of the inclusion or exclusion criteria, (2) no treatment for the enrolled lesion, (3) no data after randomization, or (4) histologically no NET or NEC after treatment completion. Additionally, a complementary analysis will be conducted using the per-protocol set (PPS), which includes participants from the FAS who have completed the allocated treatment. Adverse events will be assessed in the population of enrolled participants who have undergone the allocated treatment and have been followed up for up to 28 days after the procedure.
The primary assessment parameters, R0 resection rate in each group, and differences between the groups are analyzed. The 90% CI for the difference will be calculated using the Miettinen–Nurminen method. To assess the non-inferiority of ESMR-L, the difference between the groups will be compared to the non-inferiority margin of 8% using a Farrington–Manning test with a one-sided alpha of 0.05. If non-inferiority is demonstrated, the superiority of ESMR-L will be examined using a one-sided alpha of 0.05.
The secondary assessment parameter, en bloc resection rate, will be analyzed using the same method as the primary endpoint. The procedure time, number of hospitalization days, and total cost of the devices and agents are compared using the Wilcoxon rank-sum test. The rates of adverse events between the groups with and without closure of the resection site will be compared using Fisher’s exact test.
A subgroup analysis by expert and non-expert endoscopists will be performed for the primary endpoint, rate of adverse events, and procedure time. Comparison between expert and non-expert endoscopists will be also performed. Factors associated with R0 resection failure will be analyzed using logistic regression.
Data registration and managementThe investigators will enter anonymized data into a web-based Electronic Data Capture (EDC) system hosted at the Shizuoka Cancer Center. The EDC system and associated databases are secured and protected by passwords. A dedicated data management team (data center) established by the director of the data management office at Shizuoka Cancer Center will be responsible for various data management tasks, including managing the data randomization system, EDC system management, EDC form design, data analysis, and verification. The aggregated data will be stored semi-permanently at the datacenter. When patients request data deletion upon withdrawal of consent, their data will be deleted.
MonitoringMonitoring will be performed to ensure the safety and accuracy of the trial in accordance with the protocol. Monitoring will be conducted centrally, based on the input data collected in the EDC system at the data center. In principle, monitoring is conducted annually. The monitoring personnel will verify that this trial is conducted in compliance with the Clinical Trials Act and the study protocol.
Protocol amendmentsIn the case of the following protocol amendments, the principal investigator must notify the CRB of the amendments: (1) changes to the protocol or the informed consent document, (2) changes to the implementation plan, and (3) changes to the conflict of interest management criteria or plan. If there is a change in the implementation plan (jRCT registration content), other responsible physicians in each institution and the Minister of Health, Labor, and Welfare of Japan will be notified of protocol amendments.
CompensationThis trial will join the clinical research insurance and compensate for the following payments according to the insurance contract: (1) the amount paid by the patient for the medical expenses incurred for the treatment of health damage, (2) a certain amount other than the medical expenses required for the treatment of health damage that requires hospitalization, and (3) compensation for death or residual disability (disability levels one through three). The principal investigator will assess the causal relationship between the treatment protocol of this trial and any health problems that occur.
Dissemination policyThe progress and primary results of this trial will be disclosed on the jRCT platform. With the agreement of the principal investigator, the results of this trial will be disseminated in international peer-reviewed journals and presented at academic conferences. The confidentiality of the research participants will be strictly maintained when the results are disclosed. The datasets used and/or analyzed during the trial may be made available by the corresponding author and data center members upon reasonable request.
Comments (0)