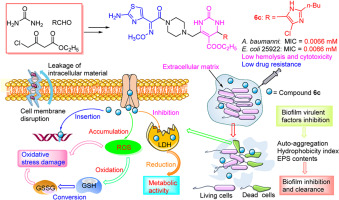
Multidrug-resistant (MDR) ESKAPE pathogens have emerged as a serious global public health threat due to the abuse of antibiotics and stalled development of new antibiotics [1]. Among these pathogens, Pseudomonas aeruginosa stands out as one of the most formidable opportunistic microbes within healthcare settings, exhibiting an escalating multidrug resistance profile and serving as a primary cause of serious pneumonia infections with mortality rates ranging from 13 % to 50 % [2]. Methicillin-resistant Staphylococcus aureus (MRSA) is also a prominent cause of hospital-acquired infection. Its biofilm can colonize indwelling medical devices such as catheters, stents, and pacemakers [3]. The mature cells within MRSA biofilm are difficult to eradicate except by surgery, while persistent cells can resume growth after treatment with high concentrations of antibiotics, leading to infection relapse and treatment failure [4].
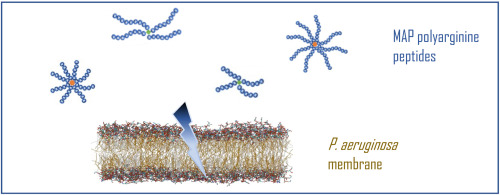
Antimicrobial peptides (AMPs) have been receiving growing recognition as promising antibacterial agents due to their ability to inhibit biofilm formation and low resistant development [5]. It primarily acts on the bacterial membrane by physical damage, evading the resistance mechanisms of conventional antibiotics [6,7]. Consequently, it exhibits exceptional bactericidal efficacy against both susceptible and MDR pathogens [7]. Notably, several AMPs have demonstrated remarkable efficacy in treating recalcitrant biofilm infections compared to conventional antibiotics [[8], [9], [10]]. LL-37, for example, inhibited biofilm formation at concentrations 16-fold lower than its minimum inhibitory concentration (MIC) against planktonic bacteria. Moreover, SAAP-148 achieved complete eradication of established biofilm-associated infections caused by MRSA and MDR Acinetobacter baumannii after a single 4-h treatment [8]. However, protease degradation remained a serious hindrance to the clinical translation of peptide antibiotics. Over the past four decades, various strategies such as cyclization [11], stapling of peptide sequences [11], incorporation of unnatural amino acids [12], and peptidomimetics [13], have been developed to alleviate/overcome this limitation, mainly focusing on prolonging the half-life and enhancing resistance towards proteases. For example, Teixobactin is a novel cyclic peptide antibiotic that has shown potent antibacterial activity against a wide range of Gram-positive pathogens and Mycobacterium tuberculosis [14,15]. It was discovered in 2015 and represented a significant breakthrough in antibiotic development. In a recent study, Qi et al. comprehensively presented the advancements of teixobactin in the areas of discovery processes, antibacterial activity, mechanisms of action, chemical synthesis, and structural optimizations. Teixobactin specifically targets the lipid II and lipid III cell wall precursors, which are essential for the growth and survival of bacteria. By binding to these molecules, teixobactin disrupts the synthesis of bacterial cell walls, ultimately leading to bacterial death [14]. Moreover, teixobactin has demonstrated remarkable effectiveness against multidrug-resistant bacteria, exhibits a low propensity for developing resistance, and maintains its serum concentration above the MIC for 4 h [16]. These inherent advantages make it a promising candidate for addressing the global health challenge of antibiotic resistance. Additionally, peptidomimetics have emerged as an increasingly appealing modification to address the limitation of peptide antibiotics in clinical applications.
Sulfono-γ-AApeptides, a novel class of peptidomimetics, possessed the limitless potential to simulate the α-helical structure of natural AMPs and introduce diverse chemical functional groups [[17], [18], [19]]. In our previous study, the introduction of sulfono-γ-AApeptides has been experimentally validated for enhancing antimicrobial activity, stability, and selectivity [20]. Compared with parent peptide, Feleucin-K3 derived peptides modified with sulfono-γ-AA exhibited a powerful and broad-spectrum antimicrobial activity, as well as potent therapeutic potential and safety in treating MDR bacterial infections [20]. However, its stability was required for further optimization [20].
In medicinal chemistry, the introduction of fluorine or fluorinated groups (such as trifluoromethyl or trifluoromethyl piperidine groups) serves as a viable strategy to overcome protease degradation. It was estimated that approximately 25 % of the drugs currently available in the market contain at least one fluorine atom [21,22]. Nowadays, fluorine is extensively utilized to improve metabolic stability and other pharmacokinetic (ADME) properties [[23], [24], [25]]. On this basis, we hypothesize that the introduction of fluorinated groups to the side chains of sulfono-γ-AApeptides may obtain novel peptides with both powerful antimicrobial activity and superior metabolic stability.
Feleucin-K3 (K3, FLKLLKKLL-NH2) was an amphiphilic cationic AMP that exhibited potent antimicrobial activity against P. aeruginosa, making it a promising candidate for antimicrobial therapy. However, its poor stability and selectivity severely limited its clinical translation [26]. The previous study extensively investigated and successfully synthesized a range of analogs, including those modified with non-natural amino acids and sulfono-γ-AA building blocks [20,[27], [28], [29]]. Here, considering the potential of fluorinated peptides to increase pharmacokinetics, we reported a novel modification strategy, featuring introducing fluorinated sulfono-γ-AA to the protease-sensitive sites of Feleucin-K3 for improving protease stability and selectivity. Benzyl and isobutyl were chosen as the R1 groups of sulfono-γ-AA, corresponding to the side chain of Phe or Leu in the parent peptide, respectively. The R2 groups incorporated highly fluorinated residues with superior metabolic stability, namely 4-(trifluoromethyl) benzene-1-sulfonyl chloride and 3,5-bis(trifluoromethyl) benzene sulfonyl chloride. A series of Feleucin-K3 analogs containing fluorinated sulfono-γ-AA were synthesized, followed by an evaluation of their antibacterial activity against both susceptible and MDR pathogens. The stability of these analogs in the presence of salt and serum was also determined, while resistant development and mechanisms were examined in detail. Finally, their antibacterial effect and toxicity in vivo were evaluated through a bacterial pneumonia/biofilm infection model and an acute toxicity test.


Comments (0)