According to the World Health Organization (WHO), stroke is the second leading cause of death worldwide, responsible for approximately 11% of all deaths. Additionally, it is also the third leading cause of disability globally [15]. From 1990 to 2019, the world experienced a 70% increase in the overall number of stroke incidents, an 85% escalation in the prevalence of strokes, and a 43% uptick in fatalities due to stroke [12]. Existing approaches to stroke prevention are inadequate, necessitating a global reinforcement of efforts to expand population-wide preventive measures. While early reperfusion is a proven technique for treating brain ischemia [33], the subsequent cerebral ischemia/reperfusion (I/R) injury that occurs following ischemic stroke and reperfusion is a complicated pathological phenomenon. Currently, cerebral I/R injury is mainly triggered by issues such as acidosis, excessive intracellular calcium, oxidative stress, inflammatory damage, imbalances in cellular energy metabolism, and FA deficiency et al. [28,42,46,48]. Given the pressing need for specific treatments, a more comprehensive grasp of the underlying pathological processes could open avenues for improved therapeutic solutions.
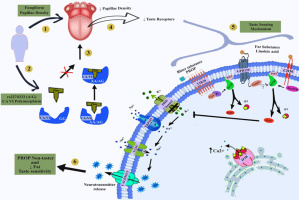
FA is a crucial nutritional element that plays a role in various metabolic processes and contributes to the functioning of a range of molecules, including purines, pyrimidines, DNA, RNA, amino acids, S-adenosyl-methionine, and serotonin [14,23]. Additionally, FA is vital for optimal brain health as it aids in the creation of nucleotides and neurotransmitters, the synthesis of myelin, and the regulation of homocysteine levels to safe concentrations. Research has shown the significance of FA in addressing numerous neurological conditions, such as neural tube defects, Alzheimer's disease, autism spectrum disorders, and stroke [7,10,26,34]. Substantial research suggests that a deficiency in FA increases vulnerability to ischemic stroke, brain damage, and neuronal harm, while supplementing with FA can lower stroke risk by 18% [10,41]. FA's natural forms are typically present in green leafy vegetables, fruits, liver, and dairy products. Since mammalian cells cannot produce FA internally, they are entirely dependent on external sources for this essential B vitamin [22]. Folate hydrolase 1 (Folh1), also known as Prostate Specific Membrane Antigen (PSMA) or Glutamate Carboxypeptidase (GCPII), is crucial for absorbing dietary FA in the digestive system. Beyond its role in the gut, GCPII is found in a variety of tissues including the nervous system, prostate, and heart. Under hypoxic conditions, there's an upregulation of GCPII expression in neuronal cells. Additionally, an increase in GCPII-positive cells has been observed in rats undergoing Middle Cerebral Artery Occlusion/Reperfusion [47]. Despite these observations, the connection between FA deficiency and elevated GCPII levels is not yet fully understood.
In the central and peripheral nervous systems, GCPII enzymatically cleaves the neurotransmitter N-acetylaspartylglutamate (NAAG), releasing Glu and consequently playing a role in various neurological disorders [2,45]. Numerous studies have shown that a rise in extracellular Glu can initiate ferroptosis, primarily by inhibiting the cystine/glutamate antiporter system (system Xc-), diminishing the activity of glutathione peroxidase 4 (Gpx4), leading to the build-up of reactive oxygen species (ROS), and reducing glutathione (GSH) levels. Therefore, GCPII has emerged as an important therapeutic focus for treating Glu-mediated disorders of the nervous system. Under pathological conditions, suppression of GCPII has demonstrated a reduction in the excitotoxicity linked to increased Glu activity.
In the study, we demonstrated that in vitro and in vivo conditions, FA deficiency prompts a transcriptional increase of GCPII as an adaptive response. This transcriptional activity is mediated through Sp1 and results in elevated levels of GCPII and Glu, thereby inducing brain ferroptosis. On the other hand, FA supplementation counteracted this brain damage by inhibiting the transcriptional activation of GCPII, consequently reducing both GCPII and Glu levels and inhibiting ferroptosis.







Comments (0)