
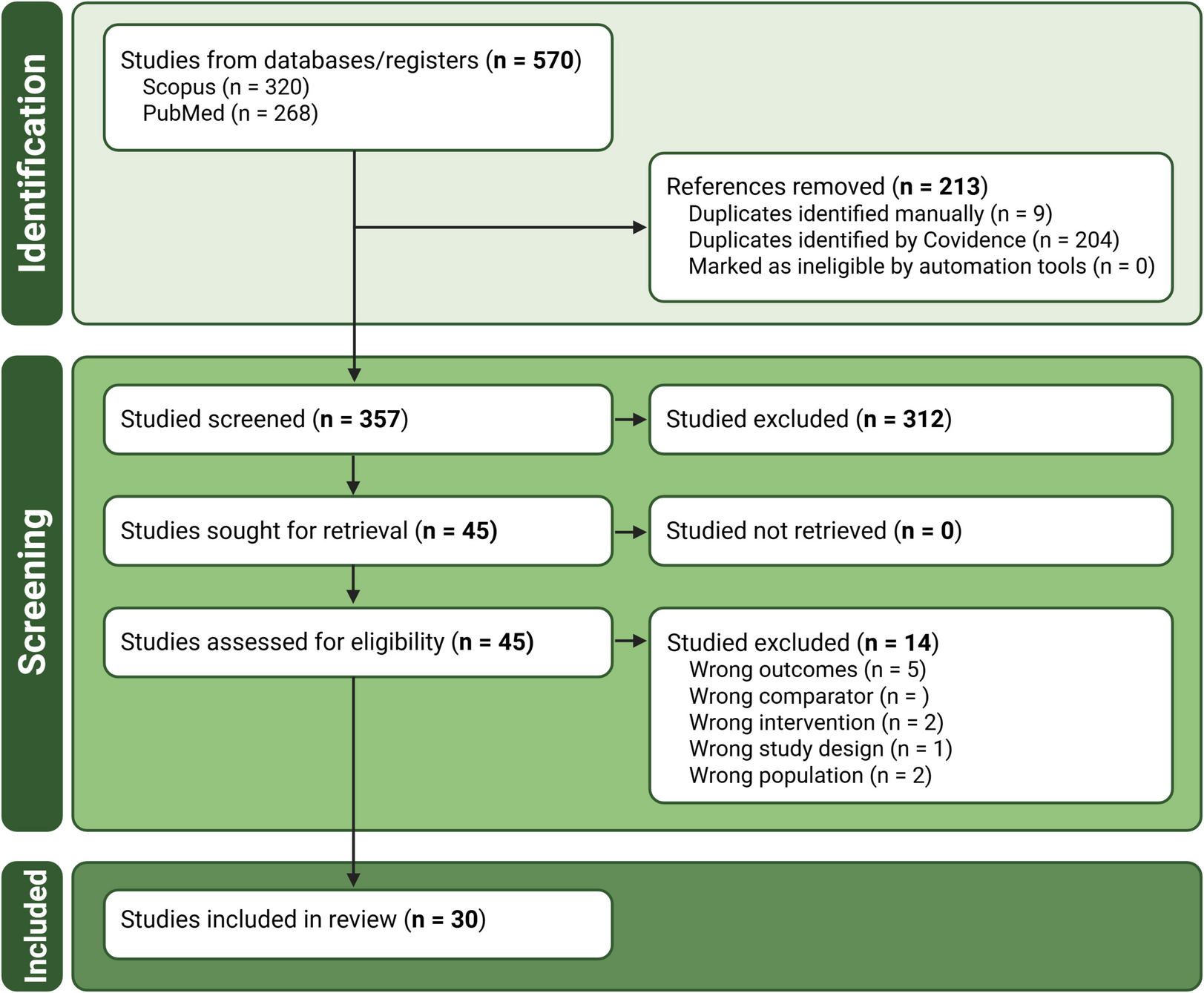
Remember me
The study consisted of five parts, as depicted in Fig. 1.
1.Bioinformatics analysis: A total of 11 mice were used in this analysis, but 1 died; thus, data were obtained from 10 mice. To investigate the pathophysiological mechanisms of brain injury following SAH, bulk RNA-seq data from mice, including the published dataset GSE79416 and our own data (sham group, n = 6; SAH group, n = 4), and human proteomics data derived from a published dataset (PXD030593) were collected for gene set enrichment analysis (GSEA). Subsequently, based on the GSEA results, the immune microenvironment, particularly the dynamic changes in the immune cell composition, was assessed using two independent techniques: mMCP_counter and ImmuCellAI-mouse.
2.Experiment 1: A total of 19 mice were used in this experiment, and 2 of these mice died. To validate the findings of the bioinformatics analysis, we induced SAH in the mice (sham group, n = 8; SAH group, n = 9). We harvested the ipsilateral cerebral hemisphere of the mice for flow cytometry and immunofluorescence analyses to investigate the changes in Ly6C-high monocytes following SAH. Additionally, we collected routine peripheral blood test data from SAH patients (n = 365) and healthy controls (n = 347) to analyze the correlations between monocytes and the development of SAH.
3.Experiment 2: A total of 46 mice were used in this experiment, and 8 of the mice died. To ascertain whether Ly6C-high monocytes originated from the bone marrow, we depleted bone marrow cells through the administration of busulfan (SAH + vehicle group, n = 6; SAH + busulfan group, n = 6). Furthermore, we established chimeric mice with bone marrow cells expressing green fluorescent protein (GFP) to trace the origin (especially the cranial bone marrow and limb bone marrow) of Ly6C-high monocytes (SAH group, n = 2; SAH + BMT group, n = 8). Additionally, we examined the proliferative activity of Ly6C-high monocytes and their precursors, including monocyte-macrophage DC progenitors (MDPs) and common monocyte progenitors (cMoPs), in both the cranial bone marrow and limb bone marrow (sham group, n = 5; SAH group, n = 5). Subsequently, we used the cell tracer CFSE to further confirm the origin of Ly6C-high monocytes (SAH group, n = 6).
4.Experiment 3: A total of 77 mice were used in this experiment, and 12 of these mice died. To investigate the infiltration mechanism of Ly6C-high monocytes, we identified significantly upregulated chemokines by analyzing bulk RNA-seq data. These chemokines were then validated by qPCR experiments (sham group, n = 6; SAH group on day 1, n = 6; SAH group on day 3, n = 6; SAH group on day 5, n = 6). To determine the cell origin of the chemokines, we separated the brain cells into CD11b-positive cells (mainly microglia) and CD11b-negative cells and detected the expression of the identified chemokines by qPCR (sham group, n = 3; SAH group, n = 4). Subsequently, we assessed the percentage of Ly6C-high monocytes in the brain by inhibiting microglia with minocycline (mino) (SAH + vehicle group, n = 5; SAH + mino group, n = 5), specifically depleting microglia in Tmem119DTR mice through the administration of diphtheria toxin (SAH + vehicle group, n = 6; SAH + DT group, n = 6), and using a CCL2 monoclonal antibody to antagonize CCL2 (αCCL2; SAH + IgG group, n = 6; SAH + αCCL2 group, n = 6).
5.Experiment 4: A total of 107 mice were used in this experiment, and 21 of the mice died. To elucidate the role of Ly6C-high monocytes in SAH, the mice were administered RS102895 to antagonize CCR2 (αCCR2), and their neurological function was assessed using the modified Garcia test, pole test, and rotarod test (SAH + vehicle group on days 1, 3, 5, and 7, n = 19, 18, 14, and 13, respectively; SAH + αCCR2 group on days 1, 3, 5, and 7, n = 19, 19, 14, and 11, respectively). Furthermore, to further investigate the mechanism by which Ly6C-high monocytes mediate neurological function recovery under SAH conditions, we evaluated the percentage and role of macrophages and monocyte-derived dendritic cells (moDCs) by flow cytometry (sham group, n = 6; SAH group on day 1, n = 6; SAH group on day 3, n = 18; SAH group on day 5, n = 12).
Fig. 1
Experimental design and study groups
MiceC57BL/6J mice (male, weighing 22–27 g, less than 5 mice per cage, total of 260 mice) were purchased from SLAC Laboratory Animal (China) and maintained under constant temperature and humidity conditions with free access to food and water.
To antagonize the effect of CCR2, C57BL/6J mice were injected intraperitoneally with RS102895 (HY-18611, MedChemExpress, USA) at 5 mg/kg immediately and 6 h after surgery and then every 12 h until sacrifice [11].
To inhibit the activation of microglia, C57BL/6J mice were injected intraperitoneally with minocycline (mino, MS0037, Maokang Biotechnology, China) at 50 mg/kg immediately and 12 h after surgery [12].
Tmem119DTR mice (male) expressing diphtheria toxin receptor (DTR) were established on a C57BL/6J background and purchased from Shanghai Model Organisms Center.
Establishment of the SAH modelThe endovascular perforation technique was used to establish the mouse model of SAH [13]. The mice were initially anesthetized with 5% isoflurane and maintained under 2% isoflurane throughout the experiment. After exposing the left carotid artery, the external and internal carotid arteries were carefully separated. The left external carotid artery (ECA) was isolated, and a blunt filament was advanced through the ECA into the internal carotid artery until resistance was encountered, which ultimately resulted in perforation of the circle of Willis. The sham mice were subjected to the same procedures except that the circle of Willis was not punctured. The mice were placed on a heating pad to maintain their body temperature during the surgery and afterward until they woke up. The severity of SAH was assessed based on the previously described SAH grading system [9] after the mice were sacrificed 24 h after the surgery (Additional file 1).
RNA extraction and qPCR analysisThe mice were deeply anesthetized and transcardially perfused with cold 0.1 M phosphate-buffered saline (PBS), and the brain tissue was harvested. For mouse brain tissue RNA extraction, total RNA was isolated with TRIzol (AG21102, Accurate Biotechnology, Hunan, Co., Ltd). Total RNA from cells separated by magnetic activated cell separation (MACS) was isolated using an RNAprep Pure Micro Kit (DP420, Tiangen, China) according to the instructions.
cDNA was synthesized using Evo M-MLV RT Premix for qPCR (AG11706, Accurate Biotechnology, Hunan, Co., Ltd.). qPCR was performed with a SYBR Green Premix Pro Taq HS qPCR Kit (AG11701, Accurate Biotechnology, Hunan, Co., Ltd.) according to the manufacturer’s protocol. The relative mRNA expression levels of the target genes were normalized to that of β-actin using the 2−ΔΔCt method. The forward and reverse primers of the genes are listed in Table 1.
Table 1 Sequences of the primersHigh-throughput RNA sequencingThe RNA amount and purity were quantified using a NanoDrop and Agilent 2100 bioanalyzer (Thermo Fisher, USA). The purified mRNA was fragmented into small pieces with fragment buffer at the appropriate temperature. First-strand cDNA was then generated by random hexamer-primed reverse transcription followed by second-strand cDNA synthesis. The cDNA fragments were then amplified by polymerase chain reaction, and the products were purified with AMPure XP Beads. The product was validated using an Agilent Technologies 2100 bioanalyzer for quality control. The double-stranded PCR products obtained in the previous step were heated, denatured and circularized based on the splint oligo sequence to obtain the final library. The final library was then amplified with phi29 to yield a DNA nanoball with more than 300 copies of one molecule. DNBs were loaded into the patterned nanoarray, and single-end 50-base reads were generated on the BGIseq500 platform (BGI-Shenzhen, China). The sequencing data were then filtered with SOAPnuke (v1.5.2), and clean reads were obtained and stored in FASTQ format. The following RNA-seq data analysis and visualization were performed in R (version 4.0.1) without special instructions.
Gene set enrichment analysis (GSEA)To determine the key biological processes that were altered after SAH, we performed a GSEA in the sham and SAH groups using our RNA-seq data and data downloaded from the Gene Expression Omnibus (GEO accession: GSE79416, mouse brain RNA-seq data, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE79416) or ProteomeXchange (PXD030593, human cerebrospinal fluid proteomics data, https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD030593) via GSEA software 4.0.1. Reference gene sets for Gene Ontology analysis (MousePath_GO_gmt.gmt) or (c5.go.bp.v7.5.1.symbols.gmt) were downloaded from the Gene Set Knowledgebase (http://www.ge-lab.org/gskb/) and Molecular Signatures Database (https://www.gsea-msigdb.org/gsea/msigdb/), respectively.
Single-sample GSEA (ssGSEA)The pathway enrichment score of each sample was calculated using the gsva function in the GSVA (gene set variation analysis) R package. “GO_BP_MM_POSITIVE_REGULATION_OF_MONOCYTE_CHEMOTAXIS” and “GO_BP_MM_MONOCYTE_CHEMOTAXIS” were downloaded from the Gene Set Knowledgebase (http://www.ge-lab.org/gskb/) to serve as reference gene sets.
Evaluation of the immune microenvironmentBecause immune cells are the major component of inflammation and immunity, we attempted to uncover the immunocyte composition based on bulk RNA-seq data using two independent techniques, mMCP_counter and ImmuCellAI-mouse [14, 15]. The results were normalized via the scale function in R to assess the consistency of the results between the two methods, and the interclass correlation coefficient (ICC) was calculated using the R package irr [16].
Immunofluorescence stainingThe mice were anesthetized by intraperitoneal injection of 1% pentobarbital sodium and transcardially perfused with cold 0.1 M PBS followed by cold 4% PFA (BL539A, Biosharp, China). After perfusion, the mouse brains were fixed in 4% PFA for 24 h and dehydrated in 30% sucrose until they sank to the bottom. The tissues were embedded in optimal cutting temperature compound and coronally sliced into 10-μm sections. The cryosections were blocked and permeabilized with blocking buffer (P0260, Beyotime, China, Cat. No.) for 1 h at room temperature. The sections were incubated with rabbit anti-CCR2 (ab273050, Abcam, UK) at 4 ℃ overnight and then with Alexa Fluor 488-conjugated secondary antibody (A-21206, Thermo Fisher, USA) for 1 h at room temperature. Subsequently, the sections were incubated with DAPI (ab104139, Abcam, UK), and Leica software was used for imaging.
Bone marrow transplantation (BMT)Male mice were injected intraperitoneally with 140 μL of 6 mg/mL busulfan (HY-B0245, MedChemExpress, USA) for the ablation of bone marrow cells at days -7, -5, and -3 before BMT. Donor bone marrow cells isolated from the hind limbs of GFP transgenic mice (R26-CAG-EGFP, male, kindly gifted by Dr. Wangwei Jing) were transferred into the recipient mice via vein injection on day 0 [17]. After 8 weeks under undisturbed conditions, the mice were used for the establishment of SAH model mice.
Intracalvarial bone marrow injectionIntracalvarial bone marrow injection was conducted as described previously [18, 19]. Briefly, the mice were fixed using a stereotaxic apparatus, the occipital and parietal bones were slightly eroded by an electric drill, and 2 μL of CFSE solution (1:5 dilution, C34554, Thermo Fisher, USA) was manually injected using a 34G Hamilton syringe to trace the immunocytes in the skull bone marrow.
Magnetic activated cell separation (MACS)After transcardiac perfusion with 0.1 M PBS, the left hemisphere was harvested and then minced and ground to prepare single-cell suspensions. The cells were filtered through a 70-μm cell strainer (WHB-70UM, WHB Biotechnology, China) and subjected to 30% Percoll (17089109, Cytiva, USA) density gradient centrifugation to remove the myelin sheath. After lysing red blood cells with 1 × RBC LYSIS BUFFER SOLN (00-4333-57, Thermo Fisher, USA), cells were incubated with CD11b microbeads (130-093-636, Miltenyi Biotec, Germany) at 4 ℃ for 15 min and then applied onto MS columns (130-042-201, Miltenyi Biotec, Germany) to collect CD11b-positive and CD11b-negative cells.
Microglia depletionFor microglia depletion, 80 ng of diphtheria toxin (DT) (0.5 ng/μL, 150, List Labs, USA) was injected intraperitoneally on day -2 and 0 before SAH induction.
Intracerebroventricular injection (i.c.v)CCL2 monoclonal antibody (16-7096-81, Thermo Fisher, USA) was used to antagonize the effect of CCL2. The mice were administered 4 μg/4 μL CCL2 monoclonal antibody or IgG control (16-4888-81, Thermo Fisher, USA) via i.c.v. injection as follows: the mice were anesthetized and fixed, a hole was drilled 0.2 mm posterior to the bregma and 1 mm lateral to the midline, and the injection was applied at a depth of 2.5 mm (1 μL/2 min). Before and after the i.c.v. injection, the needle was left in place for 5 and 10 min, respectively.
Flow cytometry analysisSingle-cell suspensions were prepared as described above. After blocking the Fc receptors with anti-CD16/CD32 antibody and assessing the live or dead status of the cells using with Zombie NIR™ Fixable Viability Kit (423106, BioLegend, USA), the cells were incubated with cell surface antibodies (Table 2) for 30 min at 4 ℃. For detection of the intracellular TNF-α and IL-10 levels, the cells were incubated with a protein transport inhibitor cocktail (00-4980-93, Thermo Fisher, USA) for 6 h at 37 ℃ before cell surface antibody staining. After fixation and permeabilization with an Intracellular Fixation and Permeabilization Buffer Set (88-8824-00, Thermo Fisher, USA), the cells were harvested and incubated with TNF-α (11-7321-81, Thermo Fisher, USA) and IL-10 (564083, Becton, Dickinson, and Company, USA) antibodies. The proliferative activity of Ly6C-high monocytes and their progenitor cells (MDPs and cMoPs) in the bone marrow was detected by Ki-67 staining. The two-chamber method was used to harvest a single-cell suspension of the hind limb bone marrow [20]; the cranium was cut into pieces and centrifuged at 500×g for 5 min to obtain a single-cell suspension. The single-cell suspensions were then filtered, stained with cell surface antibody, fixed, permeabilized with the Foxp3/Transcription Factor Staining Buffer Set (00-5523-00, Thermo Fisher, USA), and then stained with Ki-67 antibody (11-5698-82, Thermo Fisher, USA).
Table 2 Cell surface antibody used in flow cytometry analysisEvaluation of neurological functionThe Modified Garcia test includes spontaneous activity, movement symmetry of the four limbs, forelimb outstretching, climbing, body proprioception, and response to vibrissae touch, and a higher score represents better neurological performance [21]. Before modeling, the mice were subjected to pole and rotarod tests three times a day (3 days in total), and the results from the last test were recorded as the baselines. For the pole test [22], the time of the 180° turn was recorded as Tturn, and the time to descend to the bottom was recorded as Ttotal. If Tturn > 30 s, Tturn and Ttotal were both recorded as 30 s, and if the mice successfully turned within 30 s but fell or slipped or the Ttotal > 30 s, the Ttotal was recorded as 30 s. For the rotarod test [23], the initial speed of the rotating rod was 5 rpm, the acceleration was 0.2 rpm/s, and the maximum speed was 65 rpm. When the mice fell, the time and speed were recorded. The neurological function evaluation was performed by a researcher blinded to the experimental design.
Collection of clinical dataRoutine peripheral blood data of patients with aneurysmal SAH and individuals at the physical examination center (as healthy controls) were collected between May 2018 and June 2020 at the hospital. For the patients, routine peripheral blood tests were performed within 24 h after SAH. The data from patients with a history of previous stroke, infectious diseases, inflammatory diseases, or tumors, using immunosuppressive agents, and with liver, kidney, or heart dysfunction were excluded. Using the case‒control matching function in SPSS software, the patients in the SAH and healthy control groups were matched based on clinical characteristics, including age, sex, hypertension, and diabetes (Table 3).
Table 3 Clinical characteristics of the healthy controls and SAH patientsStatistical analysisThe normality and variance homogeneity of the data were analyzed based on the Shapiro‒Wilk and Levene methods, respectively. If the data met the assumptions of normality and homogeneity of variance, the significance of the data was tested by Student’s t test (two groups) or one-way analysis of variance (ANOVA) (multigroup) followed by Tukey’s post hoc test. If the data were nonnormally distributed, they were tested using the Mann–Whitney test (two groups) and Kruskal‒Wallis test (multigroup) followed by Dunn’s post hoc test. The results from the neurological function tests were analyzed by two-way ANOVA followed by the Sidak post hoc test. Typically, P values lower than 0.05 are considered to indicate significance. All statistical analyses were performed with SPSS 26 and GraphPad Prism 8.
Comments (0)