
Remember me








It may be said that efforts to define a disease are attempts to understand the very concept of the disease. This has been especially evident in systemic and primary central nervous system (CNS) vasculitides. For the past 100 years, since the first description of granulomatous angiitis of the nervous system and brain (GANS, GAB) [1] and polyarteritis nodosa (PAN) [2], they have captured the attention of generations of clinical investigators around the globe to reach a better understanding. Since my earlier review for this journal two decades ago [3], notable progress has been made in the relationship of vasculitis and headache. Primary CNS vasculitides are described in Part 2 of this article. There is a recent review of adult and pediatric systemic vasculitides [4▪▪].
 Box 1:
Box 1: no caption available
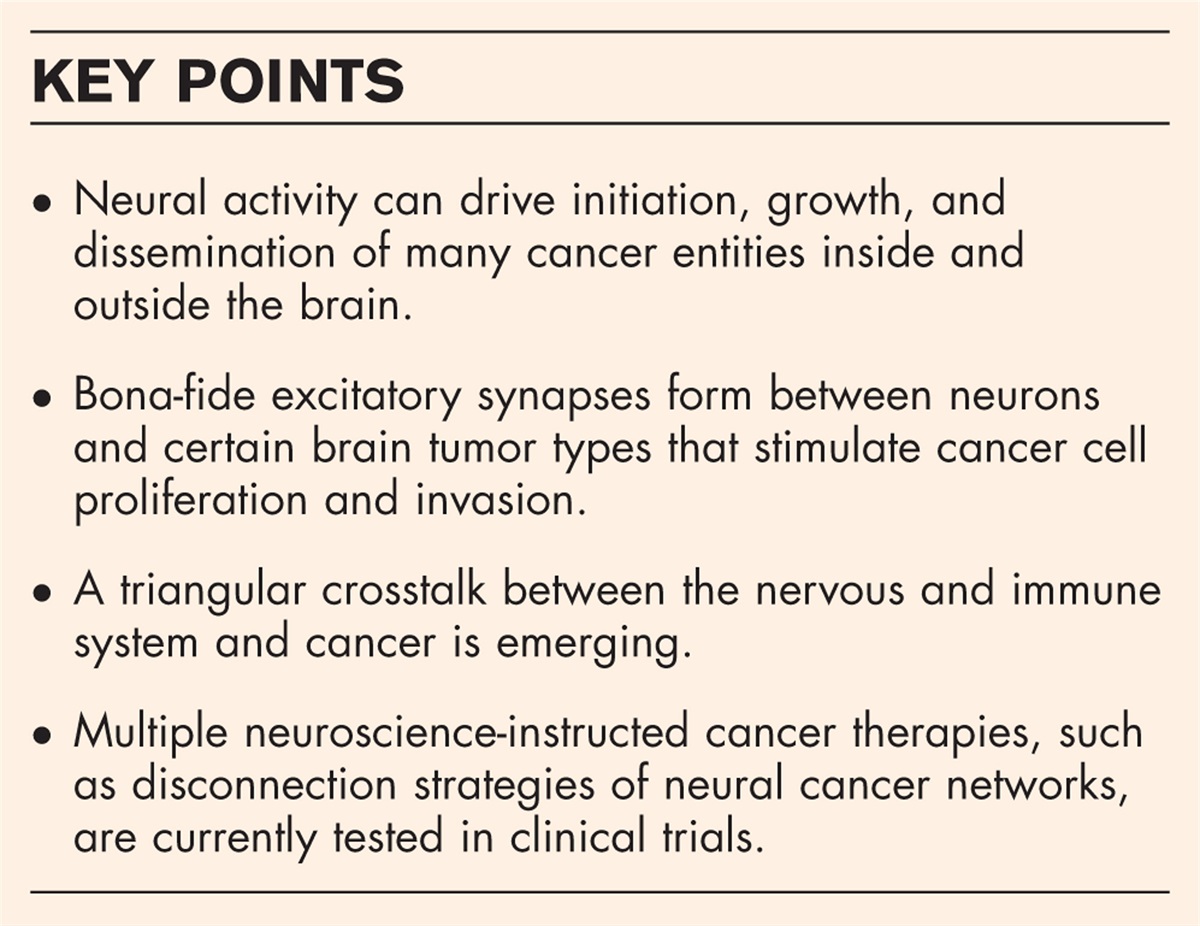
CLASSIFICATION AND NOSOLOGYThe 2012 Revised International Chapel Hill Consensus Conference (CHCC) Nomenclature of Vasculitides [5▪▪] is the most widely used classification for the vasculitides. It categorized the clinicopathologic entities based on the involved vessels and updated the nosology of the vasculitic syndromes, using specific descriptive terminology that conveyed pathophysiologic specificity (Fig. 1). The Pediatric Rheumatology European Society (PRES) and the European League against Rheumatism (EULAR) [6] in collaboration with the Pediatric Rheumatology International Trials Organization (PRINTO) reported methodology and overall clinical, laboratory and radiographic characteristics for several childhood systemic vasculitides followed by a final validated classification [7▪] also based upon vessel size, with high sensitivity and specificity [8▪].
 FIGURE 1: Diagram showing the predominant distribution of vessels affected by large vessel vasculitis, medium vessel vasculitis, and small vessel vasculitis. All categories of vasculitis can affect all types of arteries, but large vessel vasculitis most often affects large arteries. Medium arteries are most often affected by medium vessel vasculitis but large and small arteries may be affected. Small vessel vasculitis preferentially affects venules and capillaries, although arteries and veins may be affected. ANCA (antineutrophil cytoplasmic antibody) associated vasculitis affects a broad spectrum of vessels, whereas immune complex vasculitis usually affects capillaries or venules or both. The diagram depicts (from left to right) aorta, large artery, medium artery, small artery/arteriole, capillary, venule, and vein. Reproduced from [5▪▪] with permission. ANCA, antineutrophil cytoplasmic antibody; anti-GBM, anti-glomerular basement membrane.
FIGURE 1: Diagram showing the predominant distribution of vessels affected by large vessel vasculitis, medium vessel vasculitis, and small vessel vasculitis. All categories of vasculitis can affect all types of arteries, but large vessel vasculitis most often affects large arteries. Medium arteries are most often affected by medium vessel vasculitis but large and small arteries may be affected. Small vessel vasculitis preferentially affects venules and capillaries, although arteries and veins may be affected. ANCA (antineutrophil cytoplasmic antibody) associated vasculitis affects a broad spectrum of vessels, whereas immune complex vasculitis usually affects capillaries or venules or both. The diagram depicts (from left to right) aorta, large artery, medium artery, small artery/arteriole, capillary, venule, and vein. Reproduced from [5▪▪] with permission. ANCA, antineutrophil cytoplasmic antibody; anti-GBM, anti-glomerular basement membrane.The classification of vasculitides involving the nervous system with a predilection for headaches is shown in Table 1. Small vessel vasculitis (SVV) includes granulomatosis with polyangiitis (GPA) (Wegener type), microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA) [Churg–Strauss syndrome (CSS)], known collectively as antineutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAV). Vasculitic disorders associated with immune complexes (IC) include IgA vasculitis (IgAV) [Henoch–Schönlein purpura (HSP)], cryoglobulinemic vasculitis (CV or CryoVas), and hypocomplementemic urticarial vasculitis (HUV) associated with C1q antibodies. Vasculitis without a predominant vessel size and caliber, respectively from small to large, involving arteries, veins and capillaries, comprises the category of variable vessel vasculitis (VVV) characteristic of Behçet disease (BD) and Cogan syndrome. Medium vessel vasculitides (MVV) includes PAN and Kawasaki disease (KD). Large vessel vasculitides (LVV) are represented by giant cell arteritis (GCA), and Takayasu arteritis (TAK). Vascular inflammation confirmed to a single organ system such as immunoglobulin G4 (IgG4)-related aortitis [IgG4-related disease (RD)] is categorized as a single organ vasculitis (SOV).
Table 1 - Vasculitides with nervous system involvement predisposing to headaches Large vessel vasculitis Giant cell arteritis Takayasu arteritis Medium vessel vasculitis Polyarteritis nodosa Kawasaki disease Small vessel vasculitis ANCA-associated vasculitis Microscopic polyangiitis Granulomatosis with polyangiitis (Wegener) Eosinophilic granulomatosis with polyangiitis (Churg–Strauss) Immune-complex vasculitis Cryoglobulinemia IgA vasculitis (Henoch–Schönlein) Hypocomplementemic urticarial vasculitis (anti-C1q) Variable vessel vasculitis Behçet disease Cogan syndrome Single organ vasculitis Primary angiitis of the CNS Idiopathic aortitis (IgG4) Vasculitis associated with collagen vascular diseases Systemic lupus erythematosus Rheumatoid arthritisCNS, central nervous system; IgG, immunoglobulin G.
Two disorders, GCA and TAK, which fall under the category of LVV, and a third disorder, isolated aortitis, which affects large vessels, but is generally considered in the category of a SOV, can all be associated with headache.
Giant cell arteritisTwo-thirds of patients with temporal GCA present with headache, often in association with musculoskeletal complaints, in individuals of both genders and age older than 50 years [9,10]. The headache emanates along tender granulomatous lesions of inflamed extracranial vessels, including branches of the external carotid artery, including the superficial temporal, occipital, facial, and internal maxillary arteries, as well as ophthalmic, posterior ciliary, and central retinal vessels, and in the vertebral and carotid arteries to the point of dural investment. There may be tender red cords along the temple, with scalp tenderness or occipital and nuchal pain. Untreated or inadequately recognized unilateral or bilateral blindness, the result of arteritis of the intraorbital posterior ciliary and central retinal arteries, is the commonest dreaded complication, seen in up to one-half of patients. There may be oculomotor disturbances resulting from vasculitis of the extraocular muscles; vertigo and hearing impairment resulting from acute auditory artery involvement; cervical myelopathy resulting from anterior spinal artery involvement; brainstem strokes and transient ischemic attacks resulting from vasculitic involvement of the proximal intracranial carotid artery and extracranial vertebral artery.
The erythrocyte sedimentation rate (ESR) is typically increased 100 mm/h or more. Temporal artery biopsy is the only sure way of establishing the diagnosis; however, false-negative findings on the contemplated affected side may be caused by inadvertent sampling of a vasculitic-free length of vessel. Noninvasive imaging using ultrasonography, high-resolution contrast-enhanced magnetic resonance imaging (MRI), and [18F]fluorodeoxyglucose (FDG) positron emission tomography (PET) can facilitate recognition of GCA and assist the surgeon in centering on an involved segment of vessel [11]. Ultrasonography may show hypoechoic circumferential wall thickening, which occurs around the arterial lumen, termed the halo sign. Contrast-enhanced high-resolution (hr)MRI shows areas of active inflammation. [18F]FDG-PET, which can examine all of the involved vessels with a single examination, shows areas of abnormal vascular uptake typically synonymous with vessel wall inflammation.
The earliest lesions in GCA consist of vacuolization of vascular smooth muscle of the media, with enlargement of mitochondria, infiltration of lymphocytes, plasma cells, and histiocytes. Over time, inflammation extends into the intima and adventitia, leading to segmental fragmentation and necrosis of the elastic lamina, granuloma formation, and proliferation of connective tissue along the vessel wall. The classic histologic picture of granulomatous vasculitis, which is observed in about one-half of affected patients, eventuates in infiltration of vessel cell by giant cells at the junction between the intima and media, leading to thrombosis, intimal hyperplasia, and fibrosis [12].
Takayasu arteritisIndividuals younger than 50 years, particularly women of Asian descent with granulomatous arteritis affecting the aorta and its branches, are most susceptible to TAK [5▪▪]. About two-thirds of patients manifest systemic reactions at onset, including malaise, fever, stiffness of the shoulders, nausea, vomiting, night sweats, anorexia, weight loss, and irregularity of menstrual periods weeks to months before the local signs of vasculitis were recognized [13]. Headache is associated with visual loss, absent pulses in the neck and limbs with symptoms of claudication, and syncope on bending of the head backward, caused by vasculitis-related circulatory insufficiency along the aorta and branches to the brain, face, and limbs [14]. Other investigators ascribe headache to associated neck pain and carotid arterial inflammation [15] and increased propensity to migraine [16]. There may be ischemic presentations of amaurosis fugax, monocular blindness, subclavian steal and carotid sinus syndrome, audible neck and limb bruits, and asymmetry of pulses, all resulting from granulomatous vasculitis of the ascending and descending aorta and its major branches [17,18].
Although arterial biopsy is impractical given the restriction of lesions to the aorta and its branches, cerebral magnetic resonance angiography (MRA) and conventional angiography show vessel irregularities, stenosis, poststenotic dilatations, aneurysmal formation, occlusions, and increased collateralization. Although the mechanism and distribution of headache differs between patients with GCA and TAK, and pervasiveness of headache is greater in GCA than in TAK, there are strong similarities and subtle differences in the distribution of arterial disease on cerebral arteriography that suggest that the two disorders likely exist along a spectrum of the same or similar disease [19].
Immunoglobulin G4 related disorderIn 1972, Walker et al.[20] noted that 10% of 217 patients presenting with abdominal aneurysms at Manchester Royal Infirmary between 1958 and 1969 for resection showed excessive thickening of aneurysm walls and perianeurysmal adhesions at operation. Subsequent histological examination of the walls of the aneurysms showed extensive active chronic inflammatory changes including plasma-cell infiltration. In the same year, 2008, three important observations were made. First, Sakata et al.[21] concluded that inflammatory abdominal aortic aneurysm (IAAA) was related to IgG4 sclerosing disease. Second, Kasashima et al.[22] concluded that inflammatory abdominal aortic aneurysm (IAAA) was an IgG4-related disorder (RD) together with retroperitoneal fibrosis (RPF). Third, Ito et al.[23] described a patient with IAAA, hydronephrosis caused by RPF, and high levels of IgG4 in whom treatment with CS led to clinical improvement and reduction in IgG4 levels. Histological inspection of the aortic wall specimen showed lymphocytoplasmacytic infiltration. Immunohistochemical analysis of the tissue showed IgG4 positive plasma cells.
Isolated noninfectious aortitis comprises disorders characterized by chronic inflammation restricted to the aortic wall and IgG-4 infiltrating plasma cells [24]. Headache was an initial feature among 14% of patients with aortitis who had concomitant GCA or TAK [25], without which the diagnosis of coexisting aortitis might have been overlooked [26]. The clinical features of patients with inflammatory aneurysms differs from those with atherosclerotic disease due to generally younger age by a decade, lower incidence of rupture, lack of claudication of intermittent the limbs and presence of peripheral pulses, less likelihood of unusual presenting features, elevated ESR, and lack of calcification on preoperative abdominal radiographs. The risk factors for aortitis include advanced age, history of connective tissue disease, IgG4-related systemic disease (IgG4-RD), diabetes mellitus, and heart valve pathology [27]. Ultrasound and CT imaging suggests the diagnosis respectively in 13.5% and 50% of patients, the former showing a sonolucent halo with clear definition of the aortic wall posterior to the thickened anterior and lateral aortic walls.
Medium-size vessel vasculitisTwo distinct disorders, PAN and KD belong to the category of medium-vessel vasculitis, each of which can have associated headache symptoms.
Polyarteritis nodosaThere are only a few well documented postmortem series of patients with PAN to investigate the clinicopathologic correlation of the vasculitis and headache. Kernohan et al.[28] estimated that 8% of patients with PAN had CNS involvement. In a description of the postmortem findings of five pathologically studied patients with PAN, headache was nonetheless a complaint in four of them during the course of their illness, with involvement of epineurial vessels of the PNS, medium vessels of the systemic vasculature, and small meningeal arteries and large named vessels of the CNS, as follows. Patient 1 with weakness and paresthesia of the legs, and lightening pains in the limbs and head, nonetheless showed PAN involvement of epineurial vessel sparing systemic and CNS vasculature. Patient 2, who developed fatal progressive worsening of PAN with associated pain in the legs and suboccipital region, had widespread systemic PAN at postmortem examination; however, the brain was not examined. Patient 4, who complained of right supraorbital, mandibular, and aural pain, which was attributed to infected sinuses and nonerupted wisdom tooth, developed confusion, left-sided weakness and sensory loss, followed by stupor and coma before death. Postmortem examination showed near complete obstruction of the right middle cerebral artery (MCA) caused by chronic PAN involvement. Patient 5, who developed severe headache as though his head was in a vise at the onset of PAN, also died after progressive stupor and coma. Postmortem examination showed diffuse system PAN with involvement of small meningeal arteries. In these patients, headache, which was a pervasive feature at any stage of the illness, did not always correlate with the observed histopathology.
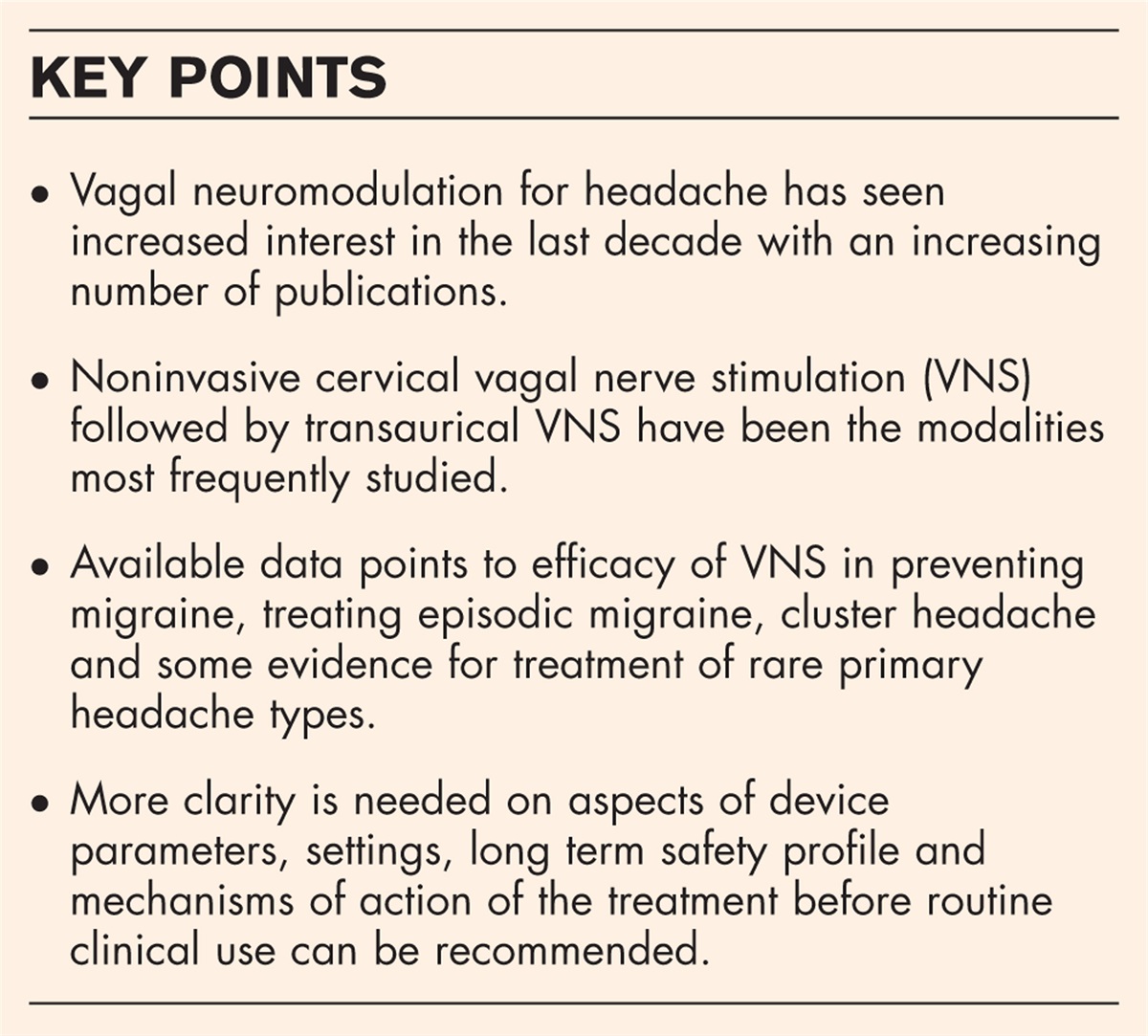
The neuropathologic changes of PAN include characteristic hyaline-like necrosis of a portion of the media and the internal elastic lamina, followed by extension of the inflammatory process to the adventitia by periarteritis (Fig. 2). The perivascular inflammation was secondary thus to the lesion in media of the artery, and although periarteritis developed, there was usually proliferation of the intima, leading to narrowing of the vessel lumen. When present, aneurysms developed during the subacute stage of the disease, leading to the gross nodosa or nodule features. Vasculitic involvement of arterioles, capillaries, and venules, and glomerulonephritis are typically absent, and there is no association with antineutrophilic cytoplasmic antibodies (ANCA), the latter of which proves to be a useful discriminatory feature.
 FIGURE 2:
FIGURE 2: Polyarteritis nodosa. This small muscular artery from muscle is from a patient with polyarteritis nodosa. There is necrosis of the media with intimal proliferation, fibrosis, and severe narrowing of the vascular lumen by dense, organized connective tissue (arrows) (hematoxylin and eosin; original magnification, ×250).
Kawasaki diseaseThis disorder was named in the honor of the investigator [29] who described acute febrile mucocutaneous syndrome with lymphoid involvement and desquamation of the fingers and toes in children. It affects medium and small arteries, particularly the coronary arteries, leading to aneurysm and ectasia formation [5▪▪]. Endothelial damage occurs in the acute stages of the illness [30]. Headache is an associated symptom, along with cough, abdominal pain, arthralgia, and seizures, which are noted in up to one-quarter of untreated patients [31]. Those with abdominal pain and headache are older by a decade or more than those without these symptoms. Migraine and Raynaud phenomenon, which coexist in some patients with KD, may be reflective of similar vascular lesions that indicate the late consequences of extra-coronary endothelial cell dysfunction [32].
SMALL-VESSEL SIZE VASCULITIS Antineutrophilic cytoplasmic antibodies -associated vasculitidesThe category of SVV includes several disorders that fall under the designations of AAV and immune complex vasculitis. The category of AAV, which is associated with necrotizing vasculitis with few or no immune deposits, predominantly affects small vessels, including capillaries, venules, arterioles, and small arteries, and is associated with myeloperoxidase (MPO) ANCA or proteinase 3 (PR3) ANCA immunocytochemistry. It includes 3 disorders: MPA, GPA, formerly termed Wegener disease, and EGPA. The other major category of SVV are the immune complex vasculitides, characterized by moderate to marked vessel wall deposits of Ig and complement components all along small arteries and veins. Immune complex SVV includes the entities of antiglomerular basement membrane disease (GBMD), CV, HUV mediated by anti-C1q antibodies, and IgAV.
Microscopic polyangiitisFever, arthralgia, purpura, hemoptysis, pulmonary hemorrhage, abdominal pain, and gastrointestinal bleeding precede the explosive phase of systemic necrotizing SVV, which affects the kidney and lungs, with rapidly progressive glomerulonephritis and pulmonary capillaritis. Cerebral signs and symptoms, including headache, was noted at presentation in 18% of patients [33]. Abnormal ANCA serology was noted in up to 80% of patients. Two of five deaths were attributed to CNS involvement by vasculitis during periods of disease at 4 and 8 months, respectively; however, that hypothesis could not be confirmed, because postmortem examinations were not performed. Microscopic polyangiitis is associated with MPO-ANCA in 58% of patients and PR3 in 26%, respectively, attributing disease activity to MPO-AAV and PR-AAV [34].
Granulomatosis with polyangiitisCNS involvement in GPA was recognized by Drachman [35] who described a patient with one month of dull bifrontal-vertex headache, which had awakened him from sleep for one month. This headache was followed by early complaints of rhinitis, nasal obstruction, epistaxis, and sensory and motor mononeuropathy multiplex, and later by disorientation, confusion, and hypertension. Many patients with GPA complain of severe constant headache attributed to destructive sinusitis early in the course of the illness. Other possible causes for concomitant headache and cerebral involvement include vasculitis of large arterial branch vessels, particularly over the surface of the brain; and hypertensive encephalopathy as suggested by microscopic infarction in the basal ganglia in close relation to arteries showing fibrinoid impregnation of their walls; and meningeal inflammation due to inflammatory cell infiltration especially plasma cells. Autoantibodies against neutrophil granule serine PR3 are detected in two-thirds of patients, and MPO in 24% of patients, respectively, attributing disease activity to PR3-AAV and MPO-AAV.
Eosinophilic granulomatosis with polyangiitisIn 1951, Churg and Strauss [36▪▪] described the clinicopathologic findings of EGPA among 13 patients with so-called allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Clinically, severe asthma, fever, and hypereosinophilia was noted, in association with widespread vascular lesions at postmortem examination, comprising fibrinoid collagen changes and granulomatous proliferation of epithelioid cells and giant cells, the so-called allergic granuloma, both within vessel walls and in connective tissue throughout the body. Other manifestations included cutaneous and subcutaneous nodules and granulomatous lymphadenitis. Contrary to the characterization that CNS involvement was rare in EGPA yet conferred a poorer prognosis [37], clinical CNS involvement, presumably headache as well, was noted in 8 (62%) of patients, varying from disorientation to convulsions and coma. Three patients with CNS involvement died of cerebral (two patients) or subarachnoid hemorrhages (one patient). Involvement of the peripheral nervous system, which is a principal criterion for the diagnosis [38] may provide a clue to the nature of the CNS lesions. Epineurial necrotizing vasculitis, noted in 54% of patients of one cohort [39] was typified by CD8+-positive suppressive/cytotoxic and CD4+-positive helper T-lymphocytes, in addition to eosinophils in inflammatory infiltrates, with only occasional CD20-positive B-lymphocytes, and scarce deposits of IgG, C3d, and IgE.
CryoglobulinemiaThe presence of one or more serum immunoglobulins that precipitate lower than core body temperatures and redissolve on rewarming is termed cryoglobulinemia [40]. Wintrobue and Buell [41] described the first patient with cryoglobulinemia, a 56-year-old woman who presented with progressive frontal headache, left face and eye pain; and right shoulder, neck, and lumbar discomfort after a bout of shingles. These symptoms were followed by Raynaud symptoms, recurrent nosebleeds, exertional dyspnea and palpitation, and changes in the eye ground attributed to central vein thrombosis and postmortem examination showed infiltrating myeloma of the humerus and lumbar vertebra, and splenic enlargement. A unique plasma protein was detected, which spontaneously precipitated with cold temperature and solubilized at high temperature and which differed from Bence-Jones proteinuria of other patients with myeloma.
Gorevic et al.[42] provided a complete description of the main clinical features and biological features of mixed CV in 40 patients, the clinical features of which included palpable purpura in all patients, polyarthralgia in three-quarters, renal disease in slightly more than half, and deposits of IgG, IgM, and complement, or renal arteritis in one-third. All cryoglobulins have rheumatoid activity consisting of IgM and polyclonal IgG, and one-third had monoclonal IgM κ components. Brouet et al.[43] provided modern classifications of cryoglobulinemia among 86 patients, which included type 1, composed of a single monoclonal immunoglobulin; and types II and III as mixed cryoglobulinemia, composed of different immunoglobulin, with a monoclonal component in type II, and polyclonal immunoglobulin in type III. In the absence of well defined disease, the presence of mixed cryoglobulinemia was termed essential.
There is a strong association with concomitant hepatitis C virus (HCV) infection and a high rate of false-negative serologic tests in type II cryoglobulinemia. The frequency of headache has not been specifically cited in any published cases of cryoglobulinemia. However, headache could be a presenting feature in those with CNS involvement, as noted in two patients with lacunar cerebral strokes and associated subcortical white matter changes on brain MRI [44,45], two patients with cortical stroke syndromes and associated cortical gray matter infarction on brain MRI [46], one patient with a temporal arteritis-like syndrome with associated ischemic cerebral infarction [47], three patients with relapsing encephalopathy [48], two patients with cerebral hemorrhage [49], two with ischemic subcortical infarcts and five with postmortem evidence of CNS involvement clinically alone or pathologically with widespread vasculitis, including the brain. With an overall mortality of 8.7%, 50 and a 33% fatality rate among those with CNS involvement [50], the symptom set of CNS involvement including headache seems to be important in prognosis.
Hypocomplementemic urticarial vasculitisThis uncommon disorder presents with recurrent attacks of erythematous, urticarial, and hemorrhagic skin lesions lasting up to 24 h at a time, associated with recurrent attacks of fever, joint swelling, and variable abdominal distress. Serum complement levels are depressed, however, immunodiffusion against purified preparations of human C1q shows strong reactivity. Skin biopsies show varied patterns of polymorphonuclear infiltration involving the vessel wall characteristic of necrotizing vasculitis, infiltration scattered diffusely through the dermis typical of anaphylactoid purpura, or mild nonspecific perivascular infiltration. Renal biopsy may show mild to moderate glomerulonephritis indistinguishable from those seen in other forms of chronic membranoproliferative glomerulonephritis. The differences in HUV from systemic lupus erythematosus (SLE) include more urticarial and purpuric skin lesions, mild or absent renal involvement, or other visceral involvement. Moreover, serum speckled antinuclear and anti-DNA antibodies, and basement membrane Ig deposits, are characteristically absent in HUV.
Among 14 patients with HUV reported by Wisnieski et al.[51], 1 patient with orbital pseudotumor complained of headache. It is unlikely that the headache in HUV would favor SLE in an individual patient, because prospective studies suggest that headache occurs in SLE at a frequency equal to normal controls [52]. Buck et al.[53] and Grotz et al.[54] cited aseptic meningitis and pseudotumor cerebri, both typified by headache, as a possible neurologic manifestation of HUV.
Immunoglobulin A vasculitis/HSPOsler [55▪] described cerebral manifestations in association with attacks of purpura in a patient with transient hemiparesis, in three others with potentially fatal hemorrhage, including one with a history of childhood attacks culminating in subdural hemorrhage, and in two others who progressed to the comatose state, one of whom had postmortem confirmation of a subdural hemorrhage; however, headache was not mentioned. Green [56] quoted a personal communication from Dr Eli Davis (St Andrew's Hospital, UK) indicating the frequency of blood in CSF in 2 of 1000 patients and reporting a child with headache and xanthochromia that followed onset of fever, malaise, sore throat, arthralgia, rash, and meningeal symptoms after presumed streptococcal illness.
The first mention of headache in this disorder was provided by Lewis and Philpott [57] in the description of three patients with neurologic complications of HSP, two of whom manifested severe headache concomitant at onset followed shortly afterward by meningeal signs and xanthochromic CSF, which indicates subarachnoid hemorrhage; a third patient without complaints of headache rapidly lapsed into coma after repeated convulsions. Postmortem examination in the only patient who died was limited to the abdominal cavity, which showed subacute nephritis and arteriolitis.
Belman et al.[58] estimated the incidence of headache to be 8.9% and noted that it was the presenting symptom in one of their three reported patients, specifically a child with a prodrome of febrile irritability, colic, nausea, and vomiting, who later developed palpable purpuric rash, hematuria, and skin biopsy, which showed leukocytoclastic vasculitis. The other two patients differed in development of other neurologic signs, which included transient postictal hemiparesis or mononeuropathy multiplex.
Recognized as a distinct entity for more than 200 years, HSP is the commonest vasculitis in children, with an incidence of 10 patients per 100 000 a year and an association with a variety of pathogens, drugs, and other environmental exposures [59]. Positive throat cultures are noted in up to one-third of cases, with group A β-hemolytic Streptococcus and titers to antistreptolysin O increased in up to one-half of cases. Recognized neurologic complications include headache, obtundation, seizures, paresis, cortical blindness, chorea, ataxia, cranial nerve palsies, peripheral neuropathy, and myositis. IgA seems to play a pivotal role in the pathogenesis of increased serum and polymeric levels of IgA [60]. IgA-containing immune complexes and rheumatoid factor [61] and selective deposition of IgA1 in glomerular mesangium in renal biopsies are present in virtually all patients with HSP nephritis and IgA nephropathy [62].
Variable-size vessel vasculitisThis category of vasculitis can affect vessels of any size, including those that are small, medium, and large, and of any type, including arteries, veins, and capillaries. Behçet disease and Cogan syndrome are two examples of a primary VVV with a propensity for vasculitis, CNS involvement, and headache.
Behçet diseaseThis disorder is characterized by relapsing aphthous ulcers of the mouth, eye, and genitalia [63]. Nervous system involvement has been estimated in 10% [64] to 25% [65] in clinicopathologically confirmed patients, with approximately one-third showing parenchymal involvement and two-thirds vascular involvement. Headache is the commonest neurologic symptom, independent of neurologic involvement in two-thirds of patients and noted to be primary in 38%, with 24% manifesting tension-type, and 15% migraines in one cohort [66]. Frontal and occipital headache and deep-seated pain around the eyes were presenting symptoms in several patients with imminent florid involvement later studied at postmortem examination [67–70] or a clue to silent neurologic involvement in other cohorts [71].
Siva and Saip [72] classified neurologic involvement into two major primary types, one caused by vascular inflammatory mechanisms with focal or multifocal parenchymal involvement, presenting most often as a subacute brainstem syndrome, and another with few symptoms and a more favorable prognosis, caused by isolated cerebral venous sinus thrombosis and intracranial hypertension. A secondary form results instead from cerebral emboli due to cardiac disease, intracranial hypertension from superior vena cava syndrome, and neurotoxicity of specific mediations used in treatment. Mortality among neurologically complicated, clinicopathologically-confirmed cases is 41%, with 59% occurring within one year of onset of neurologic involvement. Among nonfatal cases, residual neurologic signs are not uncommon.
The neuropathologic findings in BD in brain biopsies and postmortem examination have been remarkably consistent among patients over the past several decades, showing perivascular cuffing of small meningovascular and parenchymal arteries and veins [65,67,68,70,73], rarely with medium-sized arteries showing fibrinoid degeneration and recanalization, and examples of venous thrombosis [69]. The inflammatory cell infiltrates are generally comprised of lymphocytes, both T-cells and B-cells, macrophages, rarely plasma cells and eosinophils, with reactive astrocytosis and microscopic gliosis in neighboring cerebral, cerebellar, and brainstem white matter. Neuroimaging in those with neural parenchymal involvement showed a mesodiencephalic junction lesion, with edema extending along certain long tracts of the brainstem and diencephalon in 46% of patients, with the next most common location of involvement along the pontobulbar region in 40% of cases supporting a small-vessel vasculitis.
Cogan syndromeMogan and Baumgartner [74] described a 26-year-old man with recurrent headache-like pain, spasm, and redness of the left eye with photophobia, excessive tearing, and marked conjunctival injection, followed by severe attack of dizziness, tinnitus, vertigo, nausea, vomiting, ringing in the ears, profuse perspiration, and deafness. A diagnosis of recurrent interstitial keratitis (IK) and explosive Menière disease was made. In retrospect, this was probably the first reported patient with Cogan Syndrome of nonsyphilitic IK with vestibuloauditory symptoms [75]. Symptoms of IK develop abruptly and gradually resolve, associated with photophobia, lacrimation, and eye pain (which may be unilateral or bilateral), and tend to recur periodically for years, before becoming quiescent. Vestibuloauditory dysfunction is manifested by sudden onset of Menière-like attacks of nausea, vomiting, tinnitus, vertigo, and frequently, progressive hearing loss, which characteristically occurs before or after the onset of IK.
With probably fewer than 100 reported patients with this rare childhood disorder, most reported patients with typical Cogan syndrome have appeared as single case reports or patient series, often without pathologic confirmation or evidence of systemic vasculitis in a biopsy or at postmortem examination. Headache was described by Norton and Cogan [76] during the acute illness or at onset in a patient with atypical Cogan syndrome, who manifested a superior central retinal artery branch occlusion and orbital edema, as well as by Cody [77] and Cody and Williams [78] among three of 5 patients with typical Cogan syndrome and one of two patients with atypical Cogan Syndrome.
More recently, Gluth et al.[79] noted headache in 24 of 60 (40%) patients with Cogan syndrome of mean age 38 years (range 9–70 years), whereas Pagnini et al.[80] noted headache at onset in 17% of children of mean age 11 years (range 4–18 years) and in association with other systemic features, including fever, arthralgia, myalgia, arthritis, and weight loss in up to 48% of children. Haynes et al.[81] found headache less common in typical Cogan syndrome in 17% of patients at onset, compared with 27% with atypical Cogan syndrome, a finding that correlated with CNS involvement identified in 4% of patients with typical Cogan syndrome compared with 15% with atypical Cogan syndrome, respectively.
Pathologically proven necrotizing vasculitis in association with Cogan syndrome was confirmed at postmortem examination in three patients, 838 485 by examination of subcutaneous nodular tissue and amputated limbs, and postmortem examination in one patient [82] or examination of biopsy tissue alone in ten living patients [83–90]. Crawford [83] observed three patients with systemic necrotizing vasculitis, both of whom had headache at onset of Cogan syndrome. Postmortem examination in the first patient (case 1) who had frontal headaches and IK before onset of vestibuloauditory symptoms, showed necrotizing arteritis involving small arteries and arterioles of the brain, gastrointestinal tract, and kidneys, in addition to cerebral edema and petechial hemorrhages.
Single-organ vasculitisSingle-organ vasculitis affects arteries or veins of any size in a single organ without features to indicate that it is a limited expression of a systemic vasculitis [5▪▪]. Involvement of small, medium, and large vessels of a single organ can be multifocal or diffuse as in those leading to an isolated organ-related clinicopathologic syndrome of the CNS, kidneys, peripheral nerves, coronary and pulmonary vessels, and retina, or focally in the breast, genitourinary, gastrointestinal system, or aorta, particularly after incidental biopsy or surgical resection because of a related or unrelated vasculitic process [91]. The SOV IgG4-RD is considered separately in the section of LVV. Adult and pediatric primary CNS vasculitides are considered in the next article.
Vasculitis associated with systemic collagen vascular diseaseSpecific systemic disorders associated with vasculitis, and in turn with headache, include sarcoidosis and the serologically specific collagen vascular disorders such as SLE and rheumatoid arthritis (RA).
Systemic lupus erythematosusThe early concepts of the collagen vascular disorders introduced by Klemperer [92,93] stemmed from the appreciation of fibrinoid necrosis using collagen staining in patients with SLE. As collagen swells and fragments, it dissolves to form a homogeneous hyaline and granular periodic acid-Schiff-positive material. The latter fibrinoid material contains immunoglobulins, antigen–antibody complexes, complement, and fibrinogen. Organ-specific responses of the CNS of this fibrinoid material leads to recognizable clinical sequelae associated with vascular and parenchymal damage. Several fluorescent antibody tests provide serologic support of SLE. The antinuclear antibody (ANA) screen produces a homogeneous pattern in most patients, with antibodies to native double-stranded (ds) DNA and reactivity to the Smith (Sm) and ribonucleoprotein (RNP) antigens, the combination of which constitutes the extractable nuclear antigen (ENA). Circulating IgG and IgM antibodies with an affinity for charged phospholipids, antiphospholipid antibodies (APA), some of which have procoagulant activity such as the lupus anticoagulant (LAC) and the generic anticardiolipin antibody (ACL) assay using cardiolipin as the antigen probe for APA, are all important determinants of prothrombotic events, especially in the CNS, wherein there is a propensity for occlusive microangiopathy.
Borowoy et al.[94] noted a prevalence of neuropsychiatric SLE (NPSLE) of 6.4% in a cohort of 1253 patients with SLE defined by the ACR [95] compared with the reported estimates of NPSLE of 14–39% in children and adults. Headache was regarded as a nonspecific minor NPSLE manifestation of chronic disease, along with mild cognitive impairment and depression. According to Tomic-Lucic et al.[96], those with so-called late onset SLE caused by development of disease after age 50 years had a frequency of NPSLE of 6.6% compared with 36.6% in early onset disease along with a higher prevalence of comorbid conditions and higher Systemic Lupus International Collaborating Clinics/ACR damage index, despite less major organ involvement and a more benign course. Once believed to be an important cause of CNS or cerebral lupus, true vasculitis was present in only 12% of postmortem examinations in the series by Johnson and Richardson [97]. There was no mention of headache or CNS vasculitis among the 150 patients with SLE described by Estes and Christian [98] nor was there mention of headache among 50 clinicopathologic cases of SLE, one-half of whom had CNS lesions, compiled by Devinsky et al.[99]. Feinglass et al.[100] noted neuropsychiatric manifestations at onset of SLE among 3% of 140 patients compared with 37% in the course of the illness; however, headache was not specifically tabulated.
Cerebral dysfunction and headache in SLE can be caused by large vessel, small vessel involvement or both. In the series by Feinglass et al.[100], vasculitis was noted in 28% of patients, as well as in 46% of those with neuropsychiatric involvement compared with 17% of patients lacking neuropsychiatric involvement. Postmortem examination of the CNS in 10 of the 19 fatalities showed two cases of multiple large and small infarcts, one of which showed inflammatory cell infiltrates in the walls of medium-sized vessels and perivascular infiltrates around small arterioles. Although active CNS vasculitis was absent in the brain and spinal tissue of all 50 cases reported by Devinsky et al.[99], two cases nonetheless showed inactive healed CNS vasculitis suggested by focal disruption of the elastic lamina and mild intimal proliferation of a single medium-sized artery, one of which had active systemic vasculitis of the PAN type, and both of which showed Libman-Sacks endocarditis and embolic brain infarcts. Focal angiitis of the CNS with cyst-like formation around affected blood vessels was noted at postmortem in the patient described by Mintz and Fraga [101], with typical SLE rash, cutaneous vasculitis, and active neuropsychiatric involvement.
Trevor et al.[102] summarized the literature of large named cerebral vessel occlusions from 1958 to 1965 and noted one patient with an MCA stenosis progressing to occlusion and three others with angiographic internal carotid artery (ICA) occlusions, adding three new patients and suggesting a relation to the occurrence of cerebral arteritis. Two women, one age 21 years and the other age 42 years, presented with headache followed by focal neurologic symptoms attributed respectively to left MCA, followed by right ICA occlusions, and a right MCA stenosis progressing to occlusion in four months. A third patient had a left ICA occlusion without mention of headache. Johnson and Richardson [97] attributed the vasculitic nature of this process histopathologically to cerebral vasculitis mediated by acute inflammation and necrosis. Younger et al.[103] reported large named cerebral vessel occlusion attributed to circulating anticardiolipin antibodies in a young man in whom a vasculitis mechanism was not evoked.
The pathogenic mechanisms of cerebral SLE are poorly understood [104]. Immune complex–mediated vasculitis affecting small vessels is believed to account for much of the damage in CNS lupus despite the paucity of cerebral vasculitis evident in the form of inflammatory infiltrates in vessel walls at postmortem examination. In those with discrete vascular infarcts, there is a known association with the presence of circulating pathogenic antibodies, which predisposes some individuals to a high risk of stroke by both small-vessel and large-vessel occlusion [103,105].
Lupus cerebritis and meningoencephalitis are two neurologic disturbances that can be associated with preceding headache. These disturbances are noted in up to 75% of patients with SLE depending on criteria [106] and an etiopathogenesis related to antibody-mediated neuronal dysfunction is likely given the lack of correlation of symptoms of NPSLE and CNS lesions at postmortem examination, together with the transient nature of the disturbance. Patients with SLE are also predisposed to infectious episodes, including those not yet treated because of impaired B-cell function and humoral immunity, in addition to others receiving immunosuppressant medication rendered impaired in T-cell function and cell-mediated immunity [106].
Rheumatoid arthritisThe ACR and European League Against Rheumatism Collaborative Initiative [107] published classification criteria for RA; and rheumatoid vasculitis (RV) qualifies as an extra-articular manifestation of RA [108]. There remains only a slight excess mortality in patients with RV compared with RA controls after allowance for general risk factors such as age and sex [109]. Three forms of vasculitis occur in RA, affecting all calibers of blood vessels, from dermal postcapillary venules to the aorta, usually in association with circulating IgM and IgG rheumatoid factor (RF) as measured by the latex fixation test, decreased complement levels, and a positive ANA test. The first form is a proliferative endarteritis of a few organs, notably the heart, skeletal muscle, and nerves characterized by inflammatory infiltration of all layers of small arteries and arterioles, with intimal proliferation, necrosis, and thrombosis. The second form is a fulminant vasculitis indistinguishable from PAN, with less severe leukocytosis, myalgia, renal and gastrointestinal involvement, and bowel perforation. The third type takes the form of palpable purpura, arthritis, cryoglobulinemia, and low complement levels.
CNS vasculitis is rare in RV; however, the postmortem findings of nine such patients have been reported [110–117], and although not mentioned in any of them, headache would not be an unexpected feature. The duration of RA had a range of 1–30 years, with most surviving decades. The neurologic presentations included delirium, confusion, seizures, hemiparesis, Gerstmann-like syndrome, blindness, and peripheral neuropathy. Postmortem examination showed widespread systemic vasculitis in 3 patients [110,112,113].
DIAGNOSISThere is general agreement on four principles in the diagnosis of vasculitis. First, vasculitis is a potentially serious disorder, with a propensity for permanent disability, as a result of tissue ischemia and infarction; recognition of the neurologic manifestations is important in developing a differential causative diagnosis. Second, undiagnosed and untreated, the outcome of vasculitis is potentially fatal. Third, a favorable response to an empirical course of immunosuppressive and immunomodulating therapy should never be considered a substitute for the absolute proof
Comments (0)