
Remember me


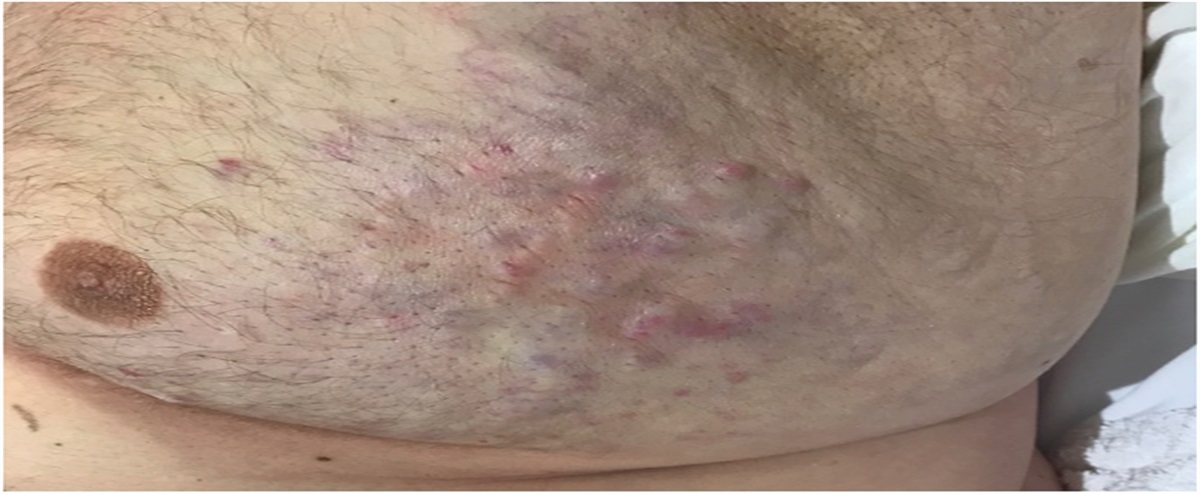
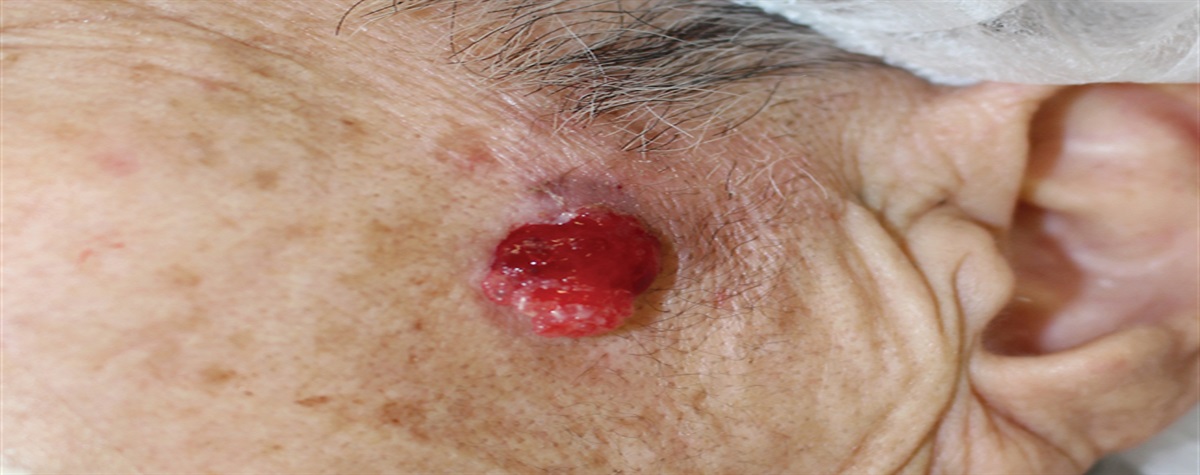
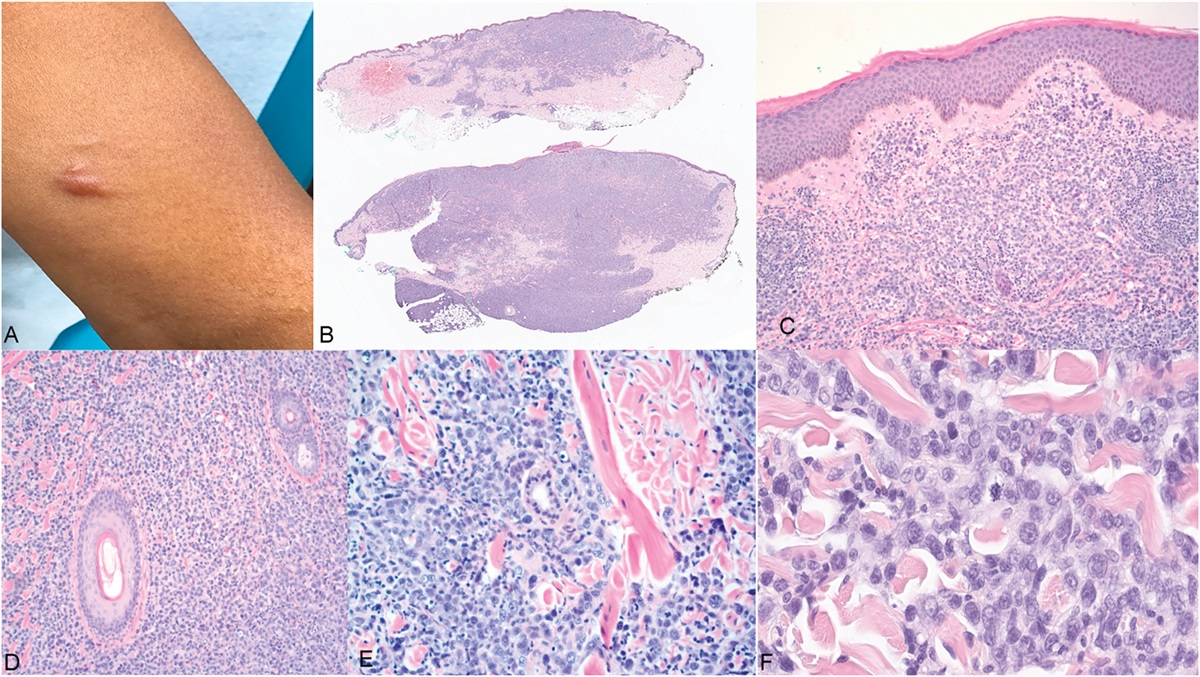
The spectrum of dermatoses defined as parapsoriasis by Brocq in 19021,2 continues to be controversial and enigmatic.3 A recent perspective on parapsoriasis has even proposed that this histopathological diagnosis should be removed from the medical dictionary as it represents cutaneous mycosis fungoides (cMF).4 Parapsoriasis can be clinically distinctive and is retained as a diagnosis by dermatologists. The enigmatic link with tumorous cMF was recognized by Bazin in 1870.5 These inflammatory dermatoses that were subsequently defined by Brocq as parapsoriasis, preceded clinically overt cMF but could also be simultaneously present and to subsequently emerge as new inflammatory lesions. A major shift, in reference to the status of parapsoriasis as a set of inflammatory dermatoses, occurred in 1979, when Sanchez and Ackerman6 defined the histopathological features of patch stage cMF. They concluded that there was no role for any subset of parapsoriasis or other primary inflammatory dermatosis to be a precursor of cMF.
Lymphomatoid papulosis (LyP) is a key in exploring the relationship of parapsoriasis and MF. In 1968, Macaulay7 defined LyP to accommodate his observation of a paradoxical combination of a self-resolving and recurring inflammatory papulonodular dermatosis that usually pursued a chronic course but was histopathologically diagnosed as cutaneous lymphoma.
Two national LyP databases of LyP8,9 with long-term follow-up of patients have indicated that either cutaneous anaplastic large cell lymphoma (cALCL) or cMF and not Hodgkin lymphoma (HL) emerged in a subset of LyP. LyP could precede, occur simultaneously, or continue to appear at times of remission of these lymphomas. This is similar to that observed in reference to clinical parapsoriasis and MF. Most patients with LyP did not develop either cMF or cALCL.
A paradigm shift in cutaneous immunodermatology can be linked with the discovery of inducible skin-associated lymphoid tissue (iSALT)10,11 hubs incorporating regulatory T cells (Tregs) that have been shown to have pleiotropic functions.12,13 These hubs have the capacity to modulate relapsing epidermotropic inflammatory and lymphomatoid dermatoses driven by overlapping immunological pathways and also control T-cell clonal expansion. This represents a central inhibiting force modulating the evolution of cutaneous T cell lymphoma (CTCL) within lymphoproliferative pathways.
In this immunological setting, LyP can be a recast as activated inducible Treg derived inhibitory lymphomatoid hubs that are inflammatory and inhibit aberrant T-cell clones in all 3 major LyP subtypes. LyP type C is recognized as a CD30+ lymphoproliferative spectrum linked to cALCL. In contrast, LyP type B relationship to cMF is more complex as CD30+ status is variable and defined by its papulonodular presentation and regression as LyP. Progression of CD30+ LyP type B is to CD30− patch/plaque form of cMF as an alternative route to parapsoriasis. Both pathways ultimately may belong to memory-based inhibitory lymphomatoid pathways that clinically appear as distinct chronic relapsing paradoxical inflammatory dermatoses linked to cMF.
In this critical analysis, LyP was the key in exploring the paradoxical relationship of parapsoriasis to MF.
Historical Perspective of LyP and CTCLCommencing in 1982, Willemze14 spearheaded detailed studies of the relationship of LyP to cutaneous and systemic lymphomas. Initially, 2 histopathological forms of LyP were distinguished: a type A, histiocytic form that could harbor Reed–Sternberg cells producing a Hodgkin-like histopathology and a type B form with an epidermotropic lymphocytic MF-like histopathology.
A significant event in the historical evolution of LyP was the emergence of the Ki-1 (later identified as CD30) monoclonal antibody derived from HL cell line.15 The application of Ki-1 antibodies led to the redefinition of the histiocytic HL-like variant of LyP as the Reed–Sternberg–like cells and also pleomorphic T cells displayed prominent Ki-1+ labelling.16 At this stage, Willemze17 noted that there was a further variant of a clinically defined LyP that showed diffuse sheets of Ki-1+ large lymphocytes. This LyP variant was to become the type C form of LyP linked to CD30+ cALCL.
The complex relationship of LyP and MF has become more apparent in later long-term studies tracking LyP to cMF.
LyP Databases Tracking the Long-Term Development of cALCL, cMF, and cHLBoth the French Study Group of Cutaneous Lymphomas and the Dutch Registry of Cutaneous Lymphoma have provided detailed results of large data-based studies of long-term monitoring of patients with LyP for progression to overt CTCL and also the risk of extracutaneous spread.
The French Study Group8 incorporated 106 patients with LyP with median follow-up duration of 11.5 years and minimum of 6 months. This study viewed all forms of LyP as biologically distinct representing a subset of cutaneous immunological inhibited CTCLs. The development of LyP could precede, occur simultaneously, or occur in remission of CTCL. A breakdown of the 3 major variants of LyP was provided, namely, cHodgkin-like LyP (type A), cMF-like LyP (type B), and cALCL-like LyP (type C). In this study, 20 cases of type A cHodgkin-like LyP were linked to 10 cases of cMF and 7 cases of cALCL, 1 case of extracutaneous MF, 2 case of extracutaneous HL, and no cases of extracutaneous ALCL. Nine cases of type B cMF-like LyP were liked to 7 cases of cMF and 1 case of cALCL, 1 case of extracutaneous HL and 2 cases of non-HL. Fifteen cases of type C cALCL-like LyP were linked to 6 cases of cALCL, 4 cases of cMF, and no cases of extracutaneous MF, ALCL, or HL but 7 cases were linked to extracutaneous non-HL.
Overall, in the French study, 38 cutaneous lymphomas comprising 26 cases of cMF and 12 cases of cALCL emerged and 14 systemic lymphomas developed in 44 of 106 patients (42%) with these 3 variants of LyP.
The Dutch study9 included 504 patients with median follow-up duration of 10 years and a minimum of 12 months, but the study did not include a breakdown of LyP subtypes. In this study, the evolution of a hematological malignancy was less than the French study with 78 of 504 LyP patients (15.5%) developing overt CTCL, including 31 cMF and 29 cALCL cases. Even after 25 years, this percentage did not exceed 20% in the Dutch study. The 31 patients with cMF that was linked to LyP were all in stage 1A and stage IB, and none progressed to tumor stage MF or extracutaneous MF.
Both studies indicated that these 3 major variants of LyP evolved to cMF and cALCL and no examples of noninvoluting cHL emerged. The Dutch study initially had 4 cases of extracutaneous HL in their data bank but significantly 2 of these were subsequently reclassified as other forms of T-cell lymphomas. In the French study, 2 cases of extracutaneous HL and 9 cases of extracutaneous non-HL emerged. These outcomes indicated that cHodgkin-like type A LyP did not transform to a non-regressing variant of cHL but was instead linked to development of either cMF or cALCL or rarely extracutaneous HL This narrow spectrum dominated by cALCL and cMF is likely to reflect linked lymphoproliferative pathways with shared T cell receptor gene rearrangement. This has been demonstrated in rare patients who have developed combinations of MF, ALCL, or HL.
Both the long-term studies indicate that all 3 subtypes of CD30+-defined LyP are linked to a lymphoproliferative spectrum that incorporate cALCL but also cMF. Even CD30+ MF-like LyP type B18 loses its CD30+ overt status when overt cMF emerges but can later undergo CD30+ large cell tumor transformation19 with decoupling of CD30+ inhibitory function. Most cases of LyP in both studies were not linked to the evolution of either cALCL or cMF, implying that the CD30+ T-cell inhibitory controls related to LyP may also apply to early MF even when not overtly expressed.
Primary cutaneous HL is currently viewed as rare. It is still possible that LyP type A is a specific variant of a specific T-cell-rich form of cHL but remains in the Treg-inhibited lymphoproliferative hubs and remains defined as HL-like LyP type A. Insights into CD30 and the tumor necrosis factor (TNF) ligand complex–related biology by Kadin20 have significance in HL and the more complex relationship of LyP and both cALCL and cMF.
The boundary of less than 75% CD30+ T cells as a histopathological criterion separating cALCL-like LyP type C from cALCL is flawed as ultimately the defining transition of lesions of LyP type C to cALCL is clinically flagged by the failure of tumors to regress.
The inhibitory nature of all major variants LyP in patients with cMF was demonstrated in the Dutch Study as patients remained in stages 1A and 1B.
The paradigm shift related to the expanding field defining the pleiotropic functional roles of iSALT Treg incorporated mesenchymal hubs was a vital element in exploring the paradoxical relationship shared both by parapsoriasis and LyP with cMF.
iSALT and Epidermotropic Mesenchymal Hubs Incorporating TregsThe concept of an immunological skin-associated lymphoid tissue comparable with the previously recognized mucosal-associated lymphoid tissue11,21 was introduced by Streilin10 in 1983. In contrast to the mucosa, the skin does not have resident lymphoid follicles. Formulating the concept of iSALT as a result of exploring immunological memory-based skin reactivity to contact sensitizers represented a pivotal advance.
The iSALT mesenchymal Treg-related complexes include keratinocytes that have been shown to produce thymopoietin-like growth factors,11,22 HLA-defined antigen processing dendritic histiocytes, and more recently defined telocytes23 governing tissue regeneration.24 CD30 has emerged as an important immunoregulator in normal T-cell development and also in the induction of CD4+/CD8+ memory T cells.25 These hubs are central to developing new committed programmed, memory-based immunological paths reacting to pathogens, allergens, or controlling oncogenic lymphomatoid dermatoses with clonally linked lymphocytic dyscrasias.
In his seminal Treg-based investigations, Rosenblum12,13 initially used ovalbumin sensitization to identify the capacity of Tregs to also transform to resident, cutaneous memory-based CD4+ Tregs (mTregs). His group has subsequently expanded and outlined the pleiotropic roles of Tregs beyond antigen-specific T-cell–based memory induction. Tregs within iSALT mesenchymal tissue have a pleiotropic range of functions that continue to be expanded (Fig. 1). These extend beyond functions that are thymus like and include the capacity to modulate and switch peripheral immunologic pathways, selectively regulate peripheral clonal T-cell expansion, activate peripheral stem cell pathways, maintain and regulate peripheral immune tolerance,26 establish peripheral adaptive memory Tregs, and modulate skin repair and inflammation. These roles have been subject of numerous studies and reviews.27–29
 FIGURE 1.:
FIGURE 1.: Wide-ranging pleiotropic functions of regulatory T cells incorporating lymphomatoid dermatoses.
This emerging immunodermatology paradigm linked to iSALT Treg hubs has particular relevance in exploring the relationship of CD30+ T-cell stem cells that define the mutated origin of cALCL and the aberrant memory CD4+/CD8+ T memory stem cells that define the mutated origin of cMF. The shared T cell receptor gene rearrangement suggest that both have a parent stem cell line, and this may be linked directly to an inducible iTreg line. The plasticity of the LyP subtypes and also immunophenotypes, particularly the expression of CD4+/CD8+ in cALCL and variable presence of CD30+ in LyP type B, may also be linked to a common iTregs origin.
The roles of Tregs within the peripheral iSALT thymus-like mesenchymal hubs can be compared with their primary immunological roles in the thymus. Particularly relevant to exploring the pathogenesis of LyP and paradoxical lymphomatoid dermatoses is the stem cell derivation of Tregs and the complex relationship of CD30+ and CD4+/CD8+ T cells.
Shared Thymus-Related Roles Incorporated in iSALT Treg-Integrated HubsTregs are derived from bone marrow progenitors that are processed in the thymus,30,31 inducing co-expression of CD4, high levels of surface CD25 (IL-2 receptor), and intracellular expression of master switch transcription factor Forkhead boxp3 (Foxp3). Negative selection occurs in the thymus as self-reacting T lymphocytes are deleted. CD4+ T cells recognizing self-antigens are simultaneously induced to express transcription factor Foxp3 that differentiate into Tregs and enforce peripheral tolerance and homeostasis. This system is finely balanced in regard to shared cutaneous anti-inflammatory autoimmune and antitumor lymphomatoid autoimmune pathways. Within the thymus, CD30/CD30L+ axis modulates T-cell proliferation, and overexpression of CD30+ helps eliminate autoreactive T cells through negative selection of CD4+/CD8+ thymocytes.20 These features that define the thymus are shared by iSALT hubs that similarly incorporate Tregs, antigen-processing dendritic histiocytes, telocytes, and Hassall corpuscle-equivalent keratinocytes within mesenchymal complexes.
To establish immune tolerance, Tregs have the now recognized capacity to modulate peripheral autoimmune pathways and defensive apoptotic systems modulating prototypic T-cell inflammatory dermatoses and oncogenic lymphomatoid dermatoses.
iSALT may have a histopathological silent but basic physiological role of regulating tissue homeostasis and cellular regeneration of tissue through multiple stem cells progenitors and rapid amplifying networks. Tregs in this aspect are key in protecting self-tolerance by modulating autoimmune forces that can disrupt homeostasis but can also assimilate genetic, immunological, and age-related mutations through the generation of pathways that acquire self-tolerance. The integration of Tregs with memory immunological-based paths can accommodate such mutations but at the expense of recurring prototypic memory T-cell–driven dermatoses.
Both MF-like/LyP and ALCL-like/LyP are expressions of focal potent Treg-derived iSALT hubs incorporating memory Treg-related regression within aberrant downstream clones of cMF and cALCL.
The most common downstream LyP Hodgkin-like subtype has inhibitory Treg hubs that can progress in a subset to either cMF or cALCL but not nonregressive cHL without the diagnostic features of either cMF or cALCL.
Reactive Lymphomatoid Pseudolymphomas and Oncogenic Lymphomatoid PathwaysReactive lymphomatoid pseudolymphomas have a histopathology that may be indistinguishable from cMF or cALCL and represent a pitfall in diagnosis and the absence of a clinical framework. MF-like lymphomatoid pseudolymphomas32 have been documented in a wide range of inflammatory dermatoses including atopic dermatitis, psoriasis, lichen sclerosus, and contact dermatitis33 but are all ultimately clinically defined as relapsing inflammatory dermatoses that do not retain their lymphomatoid histopathology. Similarly, histopathologically defined CD30+ reactive lymphomatoid cALCL-like pseudolymphomas can be seen infrequently in a wide range of clinically defined infections and infestations dominated by viruses but also more rarely in the setting of syphilis and scabies.
Both cMF and cALCL are the principal forms of CTCL linked to LyP variants that may ultimately represent upregulated iSALT Treg-integrated inhibitory lymphoproliferative hubs that are oncogenic and are shared by both lymphomas. The complex interlocked relationship of Tregs, CD30+ T cells and memory CD4+/CD8+ with iSALT peripheral thymus-like functions are shared by both reactive lymphomatoid pseudolymphomas linked to inflammatory dermatoses and the oncogenic lymphomatoid lymphomas represented by cMF and cALCL. The memory CD4+/CD8+ stem cells and CD30+ stem T cells are key members with thymus-like functions that govern T-cell clonal expansion and elimination of aberrant cells within the iSALT Treg complexes. The low incidence of 6 per million for cMF and also cALCL may reflect the potency of these homeostatic functions.
The major inhibitory hub for cMF is represented by inhibitory CD4+/CD8+ T-cell cytotoxic hub modulated by PD1/PDL-1 (programmed death 1 and ligand-1) and are expressed as lichenoid dermatoses shared by inflammatory and lymphomatoid pathways. Recurrent and prolonged lichenoid inflammation in this cytotoxic hub may result in complete resolution of individual plaques or persisting epidermal atrophy, dyspigmentation or reticulate poikiloderma forming a lymphomatoid subset linked to cMF.34
Within this framework, clinically defined parapsoriasis en plaque with the histopathology of patch stage MF but lacking the features of cMF by definition could have been classified as lymphomatoid parapsoriasis and address the more long-standing paradoxical enigma linking parpsoriasis and MF originally observed in 1870 by Bazin.
Programmed Memory Tregs Linked to Lymphomatoid cMF and Lymphomatoid cALCL Differ from Reactive Pseudolymphoma CounterpartsExperimental contact dermatitis models were used to define iSALT and also establish the induction of hapten-specific memory Tregs. Contact dermatitis can present as a reactive lymphomatoid pseudolymphomas with cMF histopathology. In this setting, the memory-based hapten pathway does not incorporate cMF. Similarly, the other cMF pseudolymphomas emerging in defined inflammatory dermatoses also do not incorporate cMF into memory-based Treg-modulated aberrant immunological pathways defining these dermatoses. In contrast, both cMF and cALCL within the context of LyP represent oncogenic lymphomatoid lymphomas.
Variants of Parapsoriasis and Their Relationship Both to Prototypic Epidermal Dermatoses and MF According to Brocq in 1902Brocq's scheme that defines parapsoriasis and their relationship to prototypic dermatoses and their links to MF (Fig. 2) represents a complex conceptual and largely clinically based template. Key morphological features defining prototypic inflammatory dermatoses such as lichen planus, psoriasis, and chronic pityriasis lichenoides (PL) had already been defined in 1902. A morphologically heterogenous group of inflammatory dermatoses also appeared in a subset of patients who developed cMF. Brocq simplified these potentially oncogenic dermatoses into 3 subtypes of parapsoriasis that morphologically differed but were interlinked to the prototypic dermatoses.
 FIGURE 2.:
FIGURE 2.: Brocq's conceptual scheme (1902) defining variants of parapsoriasis and their relationship to each other, known papulosquamous dermatoses and MF. Solid lines represent directly linked dermatoses; broad hatched bridges represent dermatoses with shared features that are not linked. Fully hatched and bridged central area with provisional lines represent complex overlapping relationship of the variants of parapsoriasis to be explored.
In his scheme, the variants of parapsoriasis are centrally placed with an outer group of prototypic inflammatory papulosquamous dermatoses. The variants of parapsoriasis were characterized by their unknown etiology, relapsing chronic state and poor response to therapy shared by the outer rim of inflammatory papulosquamous dermatoses.
In his scheme, solid lines were used to highlight prototypic dermatoses that were definitely linked to either parapsoriasis en plaque or parapsoriasis lichenoides. Broad hatched bridges were used for dermatoses, particularly psoriasis and lichen planus, which shared blended morphological features but were not directly linked to the parapsoriasis variants. There are also a complex series of shaded and dotted lines present with respect to parapsoriasis that Brocq included requiring future clarification.
Parapsoriasis en plaque and parapsoriasis lichenoides had direct transformational links to MF. Brocq also included a third guttate form of parapsoriasis that was previously defined by Juliusberg35 in 1899 as pityriasis lichenoides chronica (PLC) without a transformational oncogenic link to MF. Brocq's scheme explored the relationship of parapsoriasis to other defined inflammatory papulosquamous dermatoses. Overt clinical transformation to MF was not a defining feature for the variants of parapsoriasis. These relationships have been subsequently been the subject of several studies17,36–38 including their relationship to LyP.39,40
Critical Analysis of Brocq's Definition of Parapsoriasis and an Integrated Revised TemplateIn 1902, many inflammatory dermatoses had not been defined and those classified by Brocq as seborrheides psoriasiformes and seborrheides pityriasiques would have been heterogeneous entities (Fig. 3). Significantly, both of these potentially related seborrheic dermatoses, in contrast to lichen planus and psoriasis, were directly linked to parapsoriasis. This raises the potential that additional inflammatory variants of parapsoriasis were embedded within the spectrum of inflammatory seborrheic dermatoses. There are variants of parapsoriasis that lack the histopathogical defining features of patch stage MF such as digitate dermatosis and chronic superficial dermatitis that could have been considered clinically to represent seborrheic dermatoses. As a group, these dermatoses could be considered simply as parapsoriasis or as forms of epidermotropic T-cell dyscrasia.
 FIGURE 3.:
FIGURE 3.: Updated Brocq's template incorporating the complex web representing LyP and incorporating iSALT Treg lymphomatoid parapsoriasis as a clinically defined subset patch stage MF.
Brocq also included pityriasis rubra benin that probably incorporated pityriasis rubra pilaris (PRP) previously defined in 1857 by Devergie.41 PRP as a follicular-related dermatosis provided an opportunity to include a follicular hub without relationship to PRP. Follicular mucinosis42,43 and folliculotropic T-cell dyscrasia (folliculotropic T-cell lymphocytosis)44,45 could be included in the hub and also incorporate follicular lymphoproliferative and lymphomatoid dermatoses that now include lymphomatoid CD30+ T-cell–defined follicular LyP,46–48 CD30− folliculotropic MF including a Treg-derived stem cell basaloid folliculolymphoid hyperplastic variant of CD30− follicular MF.49,50
Regression in parapsoriasis including the lymphomatoid variant can occur without postinflammatory changes, but in chronic persistent disease, this is expressed as epidermal atrophy, dyspigmentation, and poikiloderma. Brocq's inclusion parapsoriasis lichenoides as a variant of parapsoriasis en plaque is significant as lichenoid reactions are surrogate markers for autoimmunity and are an expression of iSALT Treg-derived inhibitory pathways shared with inflammatory and lymphomatoid dermatoses.
A major challenge in the revised template was to integrate LyP within Brocq's scheme. Brocq's redefined PLC/guttate parapsoriasis proved to be the variant that could form a link to the complex LyP domain incorporating subsets including cHL-like type A that the large registry studies had shown to have the potential progress to either cALCL or cMF and was not subtype restricted.
Acute pityriasis lichenoides was defined in 1925 as pityriasis lichenoides et varioliformis acuta (PLEVA) by Mucha and Haberman.51 Macaulay had noted that PLEVA could share clinical features with LyP without a lymphomatoid histopathology. The lack of transformation of PL/guttate parapsoriasis to MF observed by Brocq has subsequently been borne out by long-term studies of PL, particularly in childhood.52 However, this lack of relationship of PL to cMF has changed as clinical variants of PL with atypical lymphomatoid histopathology53 linked to cMF have been documented. More recently, a CD30+ lymphomatoid variant of PLEVA has also been defined.54,55 These changes to the spectrum of PL also support the existence of twin lymphomatoid paths defining cMF and cALCL. The shared immunological and oncogenic Treg immunomodulatory pathways that connect CD30+ and CD4+/CD8+ T cells ultimately may define 2 oncogenic lymphomatoid domains representing LyP and lymphomatoid parapsoriasis that are now included in the revised template with their iSALT Treg affiliation.
In both cALCL and cMF, a still active iSALT Treg immunomodulatory complexes may inhibit rapid oncogenic clonal T-cell expansion and may not be distinguished by their immunophenotype but by their histopathological and cytological features and cytotoxic, apoptotic markers.
In contrast to cALCL, the CD4+/CD8+ memory stem cells that are mutated in cMF also represent the stem cells linked to the prototypic inflammatory squamoproliferative dermatoses with differing clinical and immunological profiles. This shared aspect may account for the clinical phenotypes of cMF56 that clinically resemble dermatoses such as psoriasis and atopic dermatitis and similar transitory MF-like histopathology occurring in clinical cases of these dermatoses that are classified as lymphomatoid dermatoses or pseudolymphomas. The interlinked pleiotropic functions of Tregs that include nonimmunological roles such as stem cell modulation and control of inflammatory, reparative, and regenerative pathways also have histopathological correlates. The rare stem cell–related basaloid folliculolymphoid form of folliculotropic MF may be a consequence of Tregs modulating mutated follicular stem cell pathways. Infundibulocystic and keratoacanthoma-like features have emerged in lesions of LyP, MF, and ALCL57,58 and may also be an expression of Treg modulation of reparative and oncogenic pathways linked to keratoacanthoma.59
Prototypic Inflammatory Dermatoses, Lymphomatoid Parapsoriasis, and Mycosis Fungoides and the BiologicsThe difficulties in separating prototypic inflammatory dermatoses, lymphomatoid parapsoriasis, and mycosis fungoides within their lymphoproliferative and immunological pathways is inherent to the iSALT Treg hubs and their complex integrated homeostatic, immunological, and nonimmunological functions.
This is also now reflected by the advent of biologics that have revolutionized both cancer therapy and the management of prototypic chronic relapsing dermatoses and modify iSALT Treg immunological pathways modulating inflammatory and oncogenic processes. This applies to a broad range of prototypic inflammatory dermatoses treated by biologics where the emergence lymphomatoid parapsoriasis and MF has now been documented.60 Biologics modulating TNF axis linked with CD30+ lymphocytes and checkpoint inhibitors modulating the PD-1/PDL-1 axis linked the CD4+/CD8+ cytotoxic T cells and integral to Treg-modulated pathways. This also includes folliculotropic T-cell dyscrasia presenting as a follicular eruption as a result of anti-TNF alpha therapy.61 Large numbers of lymphocytes expressing CD30 and producing Th2 cytokines have been found in acute atopic dermatitis.62,63 and CD30+ cutaneous lymphoma has emerged in association with atopic eczema.64 The same phenomenon has also occurred with duplimumab modulating interleukin-4 receptor in atopic dermatitis.65
Should Lymphomatoid Parapsoriasis Become a Synonym for a Clinically Defined Parapsoriasis Subset That has the Histopathology of Patch Stage MF?The early assignation of patch stage MF as a diagnosis provides a significant burden to patients and for parents in the case of children particularly as MF limited patch stage disease does not affect life expectancy but represents chronic relapsing dermatoses.
The adoption of lymphomatoid parapsoriasis as a synonym for a subset of parapsoriasis that lack the clinical features of cMF despite the histopathology of patch stage MF would address this issue. All presentations with the histopathology of patch stage MF including those defined clinically as cMF usually respond to current therapy but can pursue a chronic relapsing course shared by other prototypic dermatoses. The life expectancy of lymphomatoid parapsoriasis corresponds to that of cMF stages 1A and 1B and is unaltered, and no further staging procedures are currently recommended.
The preservation of parapsoriasis as a clinically based diagnosis and the incorporation of lymphomatoid parapsoriasis with the histopathology of patch stage MF may form a tribute to Brocq and his astute clinical observations. This provided a conceptual template, adopted in this critical review, to incorporate dermatological and immunological advances over a century later.
SYNOPSIS Both parapsoriasis and LyP appear clinically as inflammatory dermatoses with a paradoxical link to cMF. A key element in addressing the relationship of parapsoriasis and cMF were the results of the French and Dutch long-term registries tracking the emergence of lymphomas in the setting of LyP. Both cMF and cALCL emerged almost equally in these long-term studies but all 3 major LyP subtypes included in the registries were not specifically linked to each lymphoma. LyP Hodgkin-like variant A did not progress to cHL but instead a subset progressed to either cMF or cALCL. This ultimately supports that the distinct stem cells in both cMF and cALCL are probably derived from a common stem cell allied to inducible regulatory T cells transforming into the mutant CD4+/CD8+ memory stem cells defining cMF and mutant CD30+ stem cells defining cALCL. The question related to this shared oncogenic pathway was the immunological relationship in the skin of CD4+/CD8+ memory and CD30+ T-cell phenotypes that are expressed in both prototypic inflammatory and lymphomatoid dermatoses. This was made possible by the progress in defining iSALT mesenchymal hubs incorporating Tregs, with their pleiotropic functions defined by Rosenblum. These key skin-based domains represent a paradigm shift and formed a translational tool in this analysis. These hubs have the capacity to modulate chronic relapsing T-cell–defined inflammatory and lymphomatoid dermatoses and control T-cell clonal expansion and also modulate stem cell pathways to maintain tissue homeostasis and repair. The complex relationships that have emerged in regards to CD30+ T cells, CD4+/CD8+, and Tregs in the thymus are shared by the peripheral thymus-like iSALT hubs. Integrated CD30+ TNF/TNF-L super family complexes and CD4+/CD8+ T-cell, PD-1/PDL-1–modulated cytotoxicity within iSALT is shared by prototypic inflammatory and lymphomatoid dermatoses. These potent hubs are central to cutaneous homeostasis, peripheral adaptive tolerance, and creation of T stem cell memory pathways that define chronic relapsing prototypic inflammatory and lymphomatoid dermatoses. LyP can be recast as activated inhibitory T-cell hubs that are created by inducible iTregs in iSALT and responsible for immune surveillance shared by inflammatory and lymphomatoid dermatoses. LyP ultimately is a disorder representing inhibited oncogenesis expressed histopathologically as involuting cHL, cMF, and cALCL. The clinical course is shared by chronic relapsing inflammatory dermatoses and are adaptive inhibitory pathways that are memory based induced by the homeostatic iSALT Treg system. In 1968, LyP was defined within the context of a nondistinctive, chronic resolving and recurring papulonodular inflammatory process with a paradoxical histopathology of cutaneous lymphoma. In contrast, parapsoriasis was defined in 1902 by Brocq as a clinically distinctive spectrum of chronic relapsing lichenoid dermatoses. In 1979, Sanchez and Ackerman defined the histopathological features of early patch stage cMF in the setting of parapsoriasis and recommended abandoning parapsoriasis as these dermatoses were not simply inflammatory. This discovery could have also resulted in reclassifying parpsoriasis en plaque as lymphomatoid parapsoriasis and address the long-standing paradox and enigma related to parapsoriasis and its relationship to cMF. The oncogenic lymphomatoid presentations of cMF would now also incorporate the classic evolution cMF that is not defined by the nondistinctive relapsing papulonodular presentation shared by all 3 major stem cell–linked cutaneous T-cell lymphomas in the LyP domain. In this setting, Brocq's integration of parapsoriasis en plaque with parapsoriasis lichenoides is significant as lichenoid reactions signify antitumor immunity leading to regression and define autoimmune inflammatory dermatoses. Brocq's complex schematic chart defining parapsoriasis in relationship to known prototypic dermatoses and MF provided a template that was able to be adapted to accommodate both the lymphomatoid parapsoriasis and the LyP domains. Lichen planus and psoriasis are retained as prototypic dermatoses that are distinctive and used by Brocq to introduce the hybrid presentations of parapsoriasis. Brocq's inclusion of inflammatory guttate parapsoriasis (PLC) provided a link to the complex LyP network and an alternative but interlinked inhibitory lymphomatoid cMF subset. The CD4+/CD8+ memory stem cells defining cMF are shared within immunological pathways linked to inflammatory dermatoses that are rarely oncogenic and instead manifest as transitory cMF-like pseudolymphomas. iSALT Treg-integrated mesenchymal hubs provided an emerging translational tool that was used in redefining integrated lymphomatoid pathways. These represent activated inhibitory T-cell hubs incorporating the complex relationship bridging CD30+ and CD4+/CD8+ memory T cells incorporating thymic-related iSALT immunoregulation. REFERENCES 1. Brocq L. Les parapsoriasis. Ann Dermatol Syphilgr (Paris). 1902;35:315–322. 2. Shelley WB, Crissey JT. Jean-Lois Brocq In Classics In Clinical Dermatology: With Biographical Sketches. New York, NY: The Parthenon Publishing Group; 2003:303–316. 3. Cerroni L. The “parapsoriasis”: a riddle, wrapped in a mystery, inside an enigma. In: Cerroni L, ed. Skin Lymphoma: The Illustrated Guide. 5th ed. Oxford, UK: John Wiley & Sons Ltd; 2020:15–22. 4. Xavier JCC, Ocanha-Xavier JP, Marques MEA. Shall we exclude parapsoriasis from medical vocabulary?. J Cutan Pathol. 2021;48:833–836. 5. Bazin PAE Lecons sur le traitment des maladies chronique en general, affections de la peau en particulier. Paris: Adrein Delahaye.1870:436–438. 6. Sanchez JL, Ackerman BA. The patch stage mycosis fungoides: criteria for histologic diagnosis. Am J Dermatopathol. 1979;1:5–26. 7. Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign-histologically malignant. Arch Dermatol. 1968;97:23–30. 8. Cordel N, Tressieres B, D'Incan M, et al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76–83. 9. Melchers RC, Willemze R, Bekkenk MW, et al. Frequency and prognosis of associated malignancies in 504 patients with lymphomatoid papulosis. J Eur Acad Dermatol Venereol. 2020;34:260–266. 10. Streilin JW. Skin-associated lymphoid tissue (SALT): origins and functions. J Invest Dermatol. 1983;80(suppl l):12s–13s. 11. Ono S, Kabashima K. Novel insights into the role of immune cells in skin and inducible skin-associated lymphoid tissue (iSALT). Allergo J Int. 2015;24:170–179. 12. Rosenblum M. Skin-resident Tregs: a powerful multi-tasking force for immune balance and tissue renewals. Dermatol Focus. 2018;36:1–6. 13. Ali N, Rosenblum MD. Regulatory T cells in skin. Regul T Cell Skin Immunol. 2017;152:372–381. 14. Willemze R, Meyer CJLM, Van Vloten WA, et al. The clinical and histopathologic spectrum of lymphomatoid papulosis. Br J Dermatol. 1982;45:42–48. 15. Schwab U, Stein H, Gerdes J, et al. Production of a monoclonal antibody specific for Hodgkin and Sternberg–Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature. 1982;299:65–67. 16. Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin’s disease associated antigen Ki1in reactive and neoplastic lymphoid tissue: evidence that Reed–Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66:848–858. 17. Willemze R. Lymphomatoid papulosis: relationship to pityriasis lichenoides and malignant lymphoma. Parapsoriasis. Proceeding First International Parapsoriasis Symposium. Rochester, MN: Mayo Foundation. Edit. Muller SA; 1989:74–77. 18. Edinger JT, Clark BZ, Pucevich BE, et al. CD30 expression and proliferative fraction in nontransformed mycosis fungoides. Am J Surg Pathol. 2009;33:1860–1868. 19. Fauconneau A, Pham-Ledard A, Cappellen D, et al. Assessment of diagnostic criteria between primary cutaneous anaplastic large-cell lymphoma and CD30-rich transformed mycosis fungoides: a study of 66 cases. Br J Dermatol. 2015;172:1547–1554. 20. Kadin ME. Regulation of CD30 antigen expression and its potential significance for human disease. Am J Pathol. 2000;156:1479–1484. 21. Kogame T, Kabashima K, Egawa G. Putative immunological functions of inducible skin-associated lymphoid tissue in the context of mucosa associated lymphoid tissue. Front Immunol. 2021;12:733484. 22. Nicolas JF, Auger C, Dardenne M, et al. Do epidermal cells produce thymic hormones in vivo? An immunohistochemical study using anti-thymic hormone antibodies. Thymus. 1988-1989;12:187–201. 23. Rosa I, Marini M, Manetti M. Telocytes: an emerging component of stem cell niche microenvironment. J Histochem Cytochem. 2021;69:795–818. 24. Pittenger MF, Discher DE, Peault BM, et al. Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regen Med. 2019;4:22. 25. Zeiser R, Nguyen VH, Hou J-Z, et al. Early CD30 signaling is critical for adoptively transferred CD4+CD25+ regulatory T cells in prevention of acute graft-versus-host disease. Blood. 2007;109:2225–2233. 26. Michael M, Shimoni A, Nagler A. Regulatory T cells in allogeneic stem cell transplantation. Clin Dev Immunol. 2013;207:608951. 27. Campbell C, Rudensky A. Roles of regulatory T cells in tissue pathophysiology and metabolism. Cell Metab. 2020;31:18–25. 28. Sakaguchi S, Yamaguchi T, Nomura T, et al. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. 29. Clark RA. Resident memory T cells in human health and disease. Sci Transl Med. 2015;7:269rv1. 30. Kumar BV, Connors T, Farber DL. Human T cell development, localization and function throughout life. Immunity. 2018;48:202–213. 31. Heimal J. In: TePas E, ed. The Adaptive Cellular Immune Response: T Cells and Cytokines. 2020. Available at: https:www.uptodate.com/contents/the-adaptive-cellular-immune-response-T-cells-and cytokines. Accessed June, 2021. 32. Cerroni L. Pseudolymphoma of the skin. Skin Lymphoma: The Illustrated Guide. 5th ed. Oxford, UK: John Wiley & Sons Ltd; 2020:475–562. 33. Gomez-Orbaneja J, Iglesias Diez L, Sánchez Lozano JL, et al. Lymphomatoid contact dermatitis A syndrome produced by epicutaneous hypersensitivity with clinical features and a histopathologic picture similar to that of mycosis fungoides. Contact Dermatitis. 1976;2:139–143. 34. Lambert WC, Everett MA. The nosology of parapsoriasis. J Am Acad Dermatol. 1981;5:373–395. 35. Juliusberg F. Ube die pityriasis lichenoides chronica (psoriasiform exanthema). Archiv für Dermatologie und Syphilis. 1899;50:359–374. 36. Fortson JS, Schroeter AL, Esterly NB. Cutaneous T-cell lymphoma (parapsoriasis en plaque): an association with pityriasis lichenoides et varioliformis acuta in young children. Arch Dermatol. 1990;126:1449–1453. 37. Ko JW, Seong JY, Suh KS, et al. Pityriasis lichenoides-like mycosis fungoides in children. Br J Dermatol. 2000;142:347–352. 38. Freitag S, Boccara O, Brousse N, et al. Mycosis fungoides following pityriasis lichenoides: an exceptional event or a potential evolution? Pediatr Blood Cancer. 2012;58:307. 39. Willemze R, Scheffer E. Clinical and histologic differentiation between lymphomatoid papulosis and pityriasis lichenoides. J Am Acad Dermatol. 1985;13:418–428. 40. Wood GS, Strickler JG, Abel EA, et al. Immunohistology of pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica. J Am Acad Dermatol. 1987;16:559–570. 41. Devergie A. Pityriasis rubra pilaris. Traite Pratique des Maladies de la Peau. In: Shelley WB, Cri
Comments (0)