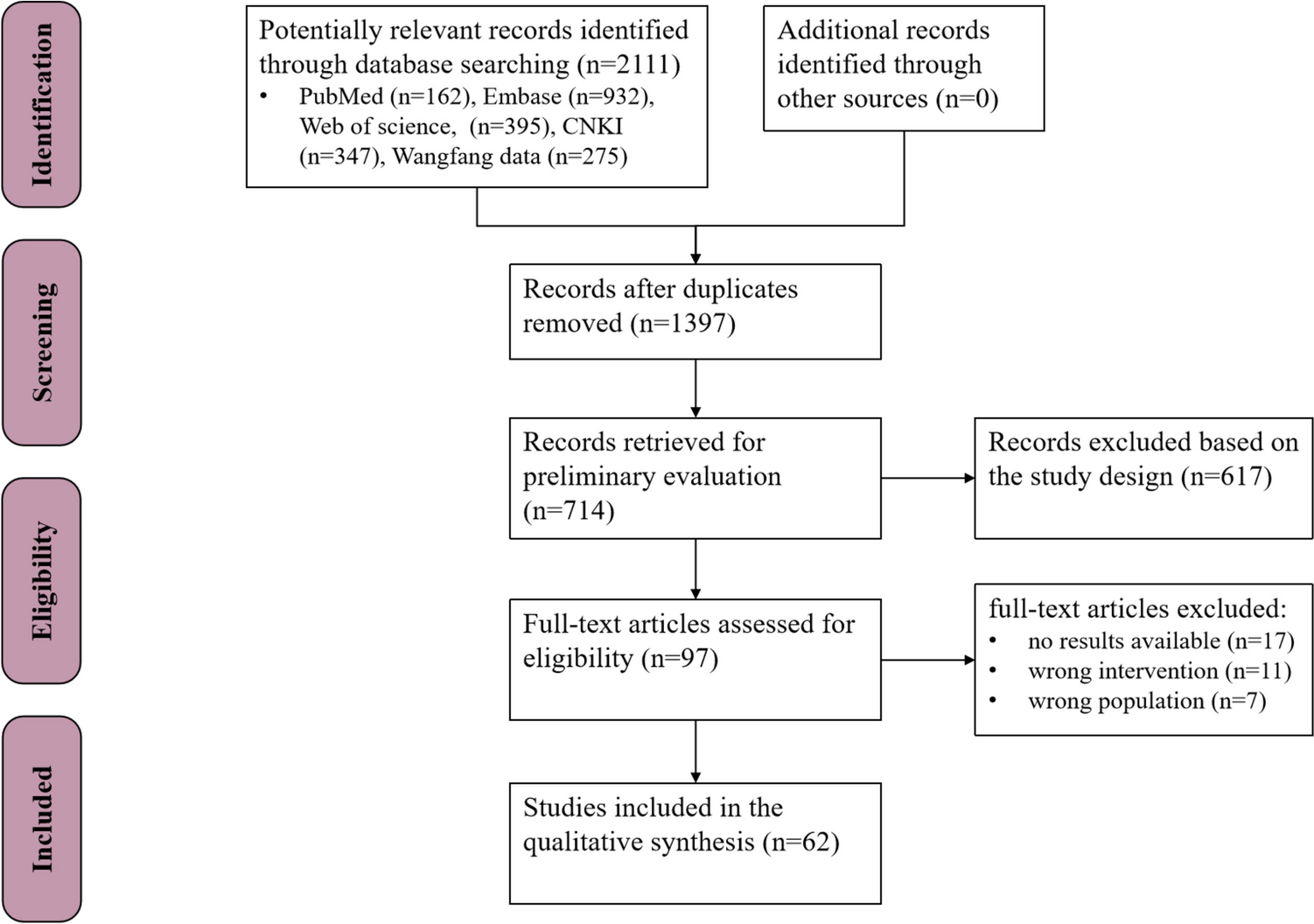
Study design
The current single-arm, prospective phase II trial was performed at the Oncology Department of Jiangsu Cancer Hospital between March 2019 and March 2022. The study followed the Declaration of Helsinki (2000), and had approval from the Ethics Committee of Jiangsu Cancer Hospital. Each patient provided signed informed consent. The current trial was registered at the Chinese clinical trial registry (www.chictr.org.cn, ChiCTR1800019421).
Participants
Inclusion criteria were: (1) 18 to 75 years old; (2) proven with ES-SCLC by histopathological examination; (3) treatment with standard first-line Etoposide-platinum solely chemotherapy, without progression; (4) Eastern Cooperative Oncology Group (ECOG) performance status of 0–1; (5) one or more computed tomography (CT) measurable lesions; (6) expected survival of at least 3 months; (7) major organ function indicators meeting the following criteria 7 days before the start of treatment: (a) hemoglobin (Hb) ≥ 90 g/L, (b) absolute neutrophil count (ANC) ≥ 1.5 × 109/L, (c) platelets (PLT) ≥ 80 × 109/L, (d) total bilirubin (TBIL) ≤ 1.5 fold the upper limit of normal range (ULN), (e) alanine (ALT) and aspartate (AST) aminotransferase levels ≤ 2.5×ULNs (ALT and AST ≤ 5×ULNs in patients with liver metastasis), (f) serum creatinine (Cr) ≤ 1.5×ULN or creatinine clearance (CCr) ≥ 60 ml/min, and (g) Doppler ultrasound evaluation showing left ventricular ejection fraction (LVEF) ≥ 50%; 7) contraceptive measures in patients of child-bearing age (female patients and female companions of male patients).
Exclusion criteria were: (1) presence of tumor types other than SCLC and mixed-SCLC; (2) a history of severe allergy or allergic constitution; (3) pregnant or breastfeeding women; (4) participation in other clinical trials; (5) pleural effusion or ascites that induced respiratory syndrome (CTCAE grade ≥ 2 dyspnea); (6) symptomatic brain metastases or symptoms controlled for < 2 months; (7) severe and/or uncontrolled diseases such as (a) suboptimal blood pressure control (systolic [SBP] and diastolic [DPB] blood pressure ≥ 150 and ≥ 100 mmHg, respectively), (b) grade ≥ 1 myocardial ischemia or infarction, arrhythmia (QTc ≥ 480 ms), or NYHA grade ≥ 2 congestive heart failure, (c) active or uncontrolled severe infection (CTCAE grade ≥ 2), (d) liver cirrhosis, decompensated liver disease, active hepatitis, or chronic hepatitis that required antiviral treatment, (e) renal failure that required dialysis, (f) a history of immunodeficiency diseases, i.e., HIV or other acquired or congenital immunodeficiency disease, or organ transplantation history, (g) suboptimal control of diabetes (fasting blood glucose (FBG) above 10 mmol/L), (h) urine routine examination showed urine protein ≥++ and 24 h urine protein above 1.0 g, or (i) neurological disease, such as epilepsy, dementia, severe depression, or mania; 8) major surgery, open biopsy, or substantial traumatic injuries within 28 days before inclusion; 9) imaging examination showing tumor invasion of the tissues surrounding vital blood vessels, or a tumor highly possibly invading vital blood vessels; 10) any grade of bleeding constitution or bleeding history, with any grade ≥ 3 bleeding or hemorrhagic events, or nonunion trauma, ulcer, or bone fracture; 11) atrial/venous embolism events within the past 6 months, including cerebrovascular events (e.g., transient ischemic attacks), deep venous thrombosis, or pulmonary embolism; 12) previous psychotropic drug abuse and incapacity of quitting, or with mental diseases; or 13) dysphagia or diagnosed drug absorption disorder.
Intervention
All patients underwent baseline imaging assessment after enrollment. The participants started treatment within 3 weeks (21 calendar days) from screening. All participants were administered anlotinib plus etoposide. Specifically, all participants were treated with oral anlotinib 12 mg q.d. on days 1–14 of 21-day cycles. Three weeks (21 d) were considered as one cycle. Anlotinib was continually administered until disease progression, consent withdrawal, or intolerable toxicity. The participants were also treated with oral etoposide 50 mg on days 1 to 14 of 21-day cycles. Etoposide treatment lasted for six cycles maximum.
During treatment, imaging examinations were performed every 2 cycles to assess clinical efficacy according to RECIST 1.1 criteria, including complete remission (CR), partial remission (PR), stable disease (SD), and progressive disease (PD). Safety were evaluated every 3 weeks (21 ± 7 days) until disease progression, consent withdrawal, loss to follow-up, or intolerable toxicity. In case of disease progression, the participants were included in the survival follow-up phase, in which follow-up was performed every 56 ± 7 days until death, loss to follow-up, or consent withdrawal. Anti-tumor therapy after disease progression was decided by the investigators. Follow-up was recommended, and patient data were recorded.
Medication was discontinued in case of disease progression. In case of grade 3 or 4 adverse effects, the oral dose of anlotinib was lowered to the next dose level. In patients using a starting dose of 12 mg/day, 10 mg/day and then 8 mg/day were subsequently used. If the initial dose was 10 mg/day, it was lowered to 8 mg/day. No dose lowering was allowed after 8 mg/d; in such cases requiring dose adjustment, treatment discontinuation was applied. When the initial dose was 8 mg/day, treatment discontinuation was directly applied if dose adjustment was necessary.
Endpoints
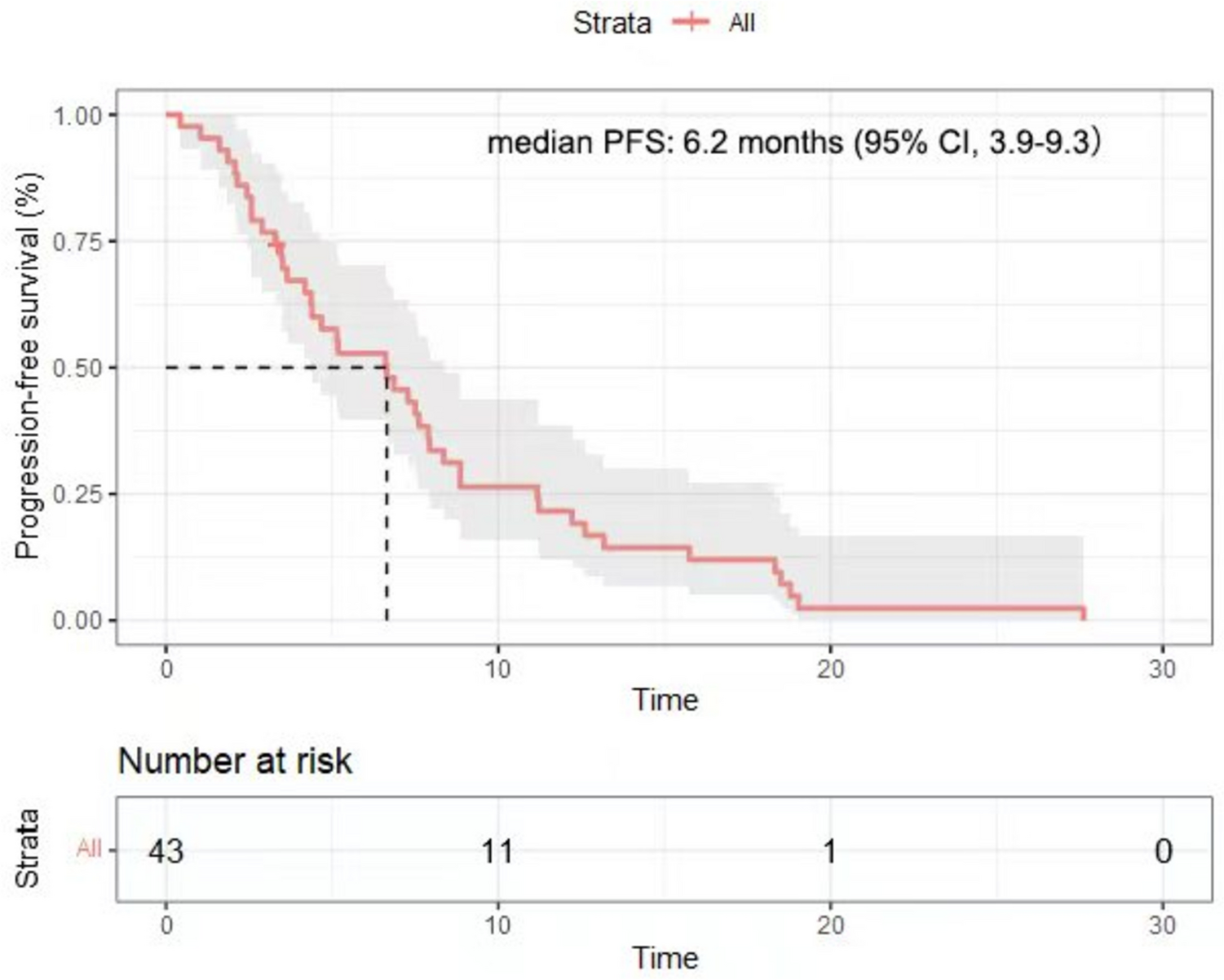
The primary endpoint was PFS, which was the time elapsed from the start of therapy to first disease progression as judged by the investigators or per imaging findings or death. Secondary endpoints encompassed OS, ORR, disease control rate (DCR), and safety. OS represented the time elapsed from the start of therapy to death. Objective Response Rate (ORR) was defined as (CR + PR) / (CR + PR + SD + PD) × 100%, and Disease Control Rate (DCR) was defined as (CR + PR + SD) / (CR + PR + SD + PD) × 100%. Treatment-emergent adverse events (TEAEs) were evaluated on the basis of the Common Terminology Criteria for Adverse Events (CTCAE) 4.0.
Sample size
Previous data revealed a PFS of 2 months in ES-SCLC without maintenance therapy following standard first-line etoposide chemotherapy [21]. The anticipated PFS of patients after anlotinib plus etoposide was 5.5 months. With α = 0.05 and β = 0.2 (power = 80%), 24 participants were required in the present trial. Considering a possible loss to follow-up of 10%, 27 participants were required.
Statistical analysis
The intent-to-treat (ITT) set encompassed all participants administered the treatment. The per-protocol analysis (PP) set included all participants with high compliance with the study protocol and strictly completed the trial processes per the study protocol. All participants in the PP set completed the drug therapy throughout the study according to the protocol.
Data were analyzed with SPSS 22.0 (SPSS, USA). Normally and skewedly distributed continuous variables were presented as mean ± standard deviation and median (range), respectively. Categorical variables were presented as n (%). Kaplan-Meier curve analysis was utilized to estimate PFS and OS.


Comments (0)