
Remember me
Primary sclerosing cholangitis (PSC) affects approximately 1 in 10,000 individuals of European ancestry. PSC is characterized by chronic immune-mediated stricturing of bile ducts, which often leads to liver failure and transplant (1,2), and is highly associated with ulcerative colitis (UC). Compared with patients with UC alone, patients with UC combined with PSC (UC-PSC) have milder but more extensive intestinal inflammation, rectal sparing, and backwash ileitis (3,4). Patients with UC-PSC are at an increased risk of disease complications including a 3- to 5-fold higher risk of colonic carcinoma than those with UC without PSC (5), frequent exacerbation of UC after liver transplant in up to 30% of patients with UC-PSC, and increased risk of pouchitis after proctocolectomy with ileoanal-pouch anastomosis (up to 90%) (6). In 84% of patients diagnosed with PSC, the diagnosis of UC is made before or at the same time as that of PSC (1). It is important for clinicians to identify which patients with UC are more likely to have or develop PSC because PSC could considerably influence UC disease behavior and clinical outcomes (eg, dysplasia, colorectal cancer). In addition, knowing the risk of developing PSC would allow clinicians to personalize the therapeutic approach.
The etiology of PSC has not been clearly identified; however, studies of heritability have indicated a strong genetic component. The relative risk (RR) of PSC developing in siblings of a diagnosed patient is higher (RR, approximately 100) (7) than in those with UC (RR, 6–9) (8). Previous genome-wide association studies (GWAS) suggest the presence of shared genetic risk variants between PSC and UC (6,7,9) in 23 regions of the genome, including the HLA region at chromosome 6p21 (9–12). A recent multiethnic meta-analysis of GWAS, which included European and East Asian cohorts, revealed another 20 novel loci involving candidate genes FCRL3, INAVA, PRDM1, IRF7, CCR6, CD226, and IL12RB1, which play key roles in immunity (13). Among established PSC loci, several have been found associated with UC (6p21, 3p21, 2q35, interleukin 21/22, caspase-recruitment domain family member 9 [CARD9], v-rel reticuloendotheliosis viral oncogene homolog [REL], BCL2L11, and UBASH3A) (9,14); although, recent evidence suggests that comorbid PSC and UC are probably the result of a unique disease genetically distinct from classical UC (10).
However, genetic biomarkers differentiating patients with UC-PSC from those with classical UC remain poorly defined. In this study, we hypothesized that unrevealed unique genetic loci exist. Through a GWAS approach, we used a large discovery cohort and an independent replicated cohort to develop and replicate novel genetic variants that differentiate patients with UC-PSC from those with UC alone.
MATERIALS AND METHODS GWAS datasetsMulticenter GWAS datasets were used for this study. The combined Mayo Clinic and Washington University IBD GWAS cohorts (Illumina Immunochip custom genotyping array) served as the discovery dataset, and the Cleveland Clinic IBD GWAS cohort (Illumina HumanOmni1-Quad BeadChip) served as the replicate data set. All data collection and study procedures were approved by the institutional review board of each cohort's respective institution.
All GWAS cohorts consisted of adult patients (aged 18 years or older) with a confirmed diagnosis of IBD. Clinical phenotype information was obtained through chart review, and blood samples were collected for DNA sequencing. All participants gave written informed consent. Clinical variables included sex, age at diagnosis of UC, disease duration, disease location, smoking history, family history of IBD, surgical resection of intestine, and extraintestinal manifestations. Extraintestinal manifestations, including joint involvement (small or large joints, ankylosing spondylitis, and sacroiliitis), eye involvement (iritis, uveitis, scleritis), skin involvement (erythema nodosum, pyoderma gangrenosum), and PSC, were retrieved from chart review. Disease location, extent, and severity were classified according to the validated National Institute of Diabetes and Digestive and Kidney Diseases Inflammatory Bowel Disease Genetics Consortium modification of the Montreal classification, as previously described (15). For this study, we limited the study population to patients who have been diagnosed with UC. Patients diagnosed with Crohn's disease or indeterminate colitis were excluded from this study.
Quality control criteriaQuality control criteria were applied for single-nucleotide polymorphisms (SNPs): (i) Hardy-Weinberg equilibrium test P value > 1.0E-05; (ii) minor allele frequency ≥0.01; and (iii) call rate ≥95%. In total, 213,386 SNPs passed quality control criteria. Samples with cryptic relatedness (estimated identical-by-descent, PI-HAT >0.25) were excluded. As a data quality assurance step, linkage disequilibrium (LD) pruning inactivated markers that were in LD with other markers, allowing us to test directly on a set of activated markers representative of the genetic information. For markers in high LD, we used pairwise expectation-maximization algorithm–based LD calculation to perform the LD pruning (thresholds: R2 = 0.50, window size = 50, and increment = 5).
Statistical analysisFor genetic association analysis with UC-PSC risk, a 2-step approach was applied: (i) SNPs that reached P < 1.00E-04 in the discovery cohort were further validated in the replicate cohort; (ii) SNPs that reached P < 0.05 in the replicate cohort were considered validated. Principal components were computed in the datasets and the first 3 principal components were included in the logistic regression models for adjusting population stratification. To estimate single locus effects for risk alleles and genotypes, we calculated odds ratios (ORs) and 95% confidence intervals (CIs) Two-sample t tests were used for continuous variables, and nominal data were analyzed by using the χ2 test or Fisher exact test. Based on sensitivity analysis at different cutoff levels, age at diagnosis was analyzed as a continuous variable and further analyzed as a nominal variable (younger than 20 years vs 20 years or older). A 2-tailed P < 0.05 was considered significant.
Model predictabilityThe odds of developing PSC were estimated through multivariate logistic regression analysis in BlueSky Statistics 7.40. The final predictive model was determined through the forward stepwise selection process. The proportion of disease variation explained by the model was estimated with McFadden pseudo R2 (16) provided by logistic regression analysis in BlueSky Statistics 7.40. Multivariate logistic regression models derived from clinical and genetic predictors were used to investigate the predictive accuracy of models predicting PSC risk in patients with UC. The discriminative accuracy of models was estimated by using area under the receiver operating characteristic curves (AUCs). AUCs were compared between models of clinical predictors only and combined clinical and genetic predictors using likelihood ratio tests.
RESULTS Clinical predictors associated with patients with UC-PSCAmong 581 patients of European ancestry with UC (discovery cohort, n = 381 [65.6%]; replicate cohort, n = 200 [34.4%]) who were successfully genotyped and had available detailed clinical information, 57 (9.8%) were diagnosed with PSC (discovery cohort, n = 46 [12.1%]; replicate cohort, n = 11 [5.5%]).
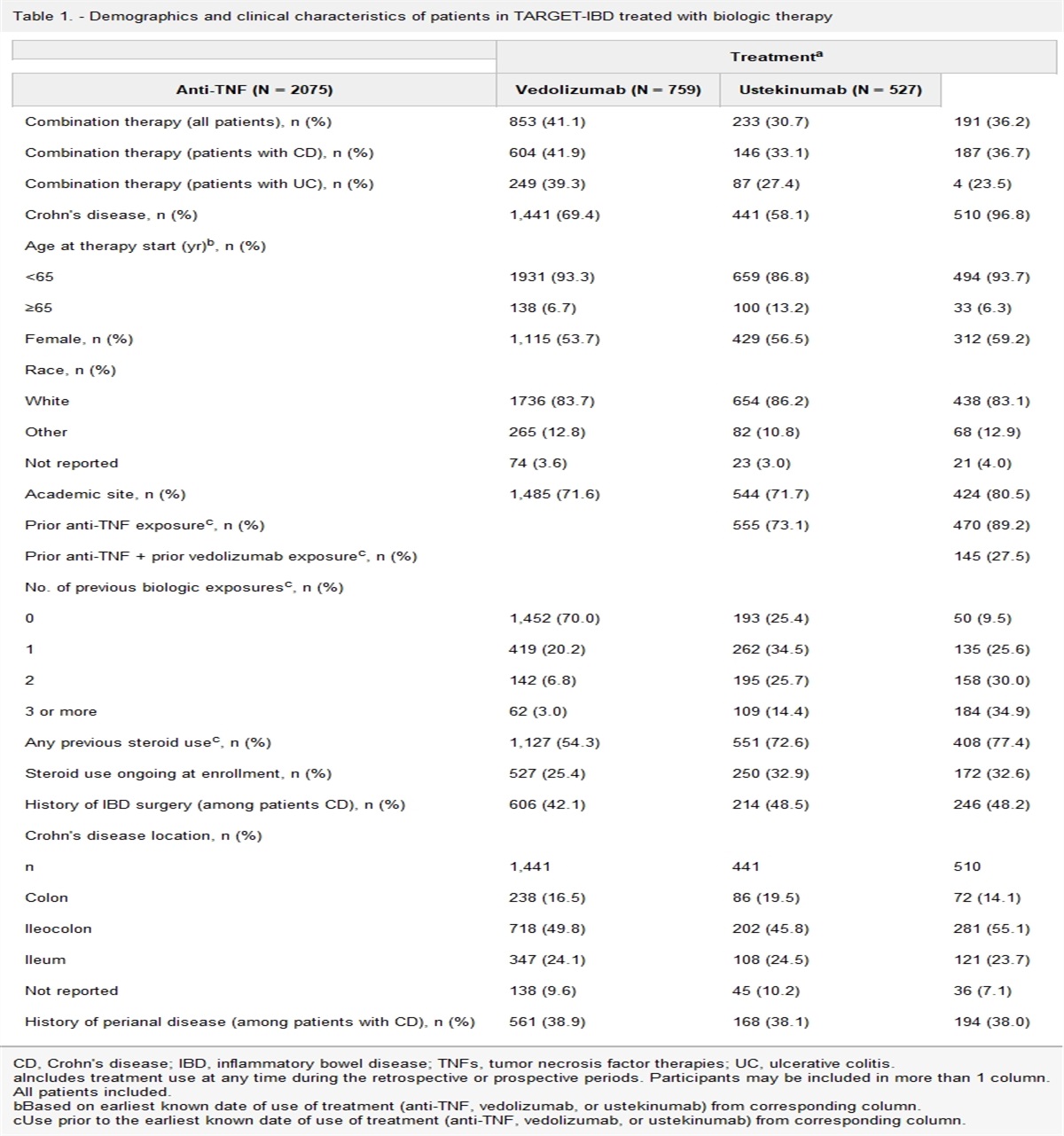
In univariate analysis of the combined dataset, compared with patients with UC alone, patients with UC-PSC were younger during diagnosis of UC (24.8 vs 33.6 years, P = 1.01E-06), had a lower rate of history of smoking (14% vs 30%; OR, 0.38 [95% CI, 0.18–0.82]; P = 0.007), had a longer disease duration (21.8 vs 11.7 years, P = 1.76E-11), and had more extensive disease (pancolitis) (93% vs 68%; OR, 5.97 [95% CI, 2.12–16.7]; P = 1.92E-05) (Table 1). Both groups were similar in terms of sex, family history of IBD, bowel resection, and extraintestinal manifestations.
Table 1. - Clinical and demographic predictors of patients with UC-PSC and UC without PSC Clinical/demographic predictors UC-PSC (n = 57) UC_without PSC (n = 524) Univariate OR (95% CI) Multivariate OR (95% CI)a Male, No. (%) 32/53 (60.3) 264 (50.4) 1.43 (0.81–2.52)IBD, inflammatory bowel disease; NA, not available; OR, odds ratio; PSC, primary sclerosing cholangitis; UC, ulcerative colitis.
aMultivariate analysis included age at UC, diagnosis, disease duration, ever smoking, and extensive disease (pancolitis).
Multivariate analysis showed patients with UC-PSC, compared with those with UC alone, had younger age at diagnosis (younger than 20 years; OR, 2.22 [95% CI, 1.16–4.25]; P = .02), longer disease duration (P = 4.42E-09), less smoking (OR, 0.42 [95% CI, 0.19–0.96]; P = 0.02), and more extensive disease (pancolitis) (OR, 5.42 [95% CI, 1.87–15.7]; P = 1.57E-04) (Table 1). These results remained significant after adding the genetic variants (rs3131621, rs9275596, rs11244), except age at diagnosis (younger than 20 years; OR, 1.67 [95% CI, 0.82–3.41]; P = 0.16) (see Supplementary Table 1, https://links.lww.com/CTG/A974).
Genetic variants associated with patients with UC-PSCIn the discovery cohort, 371 SNPs reached genome-wide significance level (P < 5.00E-08) and 1,031 SNPs reached suggestive evidence level (P < 1.00E-04). Among these 1,031 SNPs, 425 were found to be significant in the replicate cohort (nominal P < .05). These SNPs were in a high-LD 6p21.3 region. After LD pruning, 20 SNPs remained. Through a forward stepwise selection logistic regression model (selection threshold P < .01), 3 SNPs remained (rs3131621, rs9275596, rs11244) (Table 2).
Table 2. - Genetic variants associated with PSC risk in patients with UC Genetic variants: (minor/major allele) position (GRCh38.p13)GRCh38, Genome Reference Consortium Build 38; NA, not available; OR, odds ratio; PSC, primary sclerosing cholangitis; SNP ID, single-nucleotide polymorphism identification; UC, ulcerative colitis; MAF, minor allele frequency.
aAdditive genetic mode.
bMultivariate analysis included age at UC, diagnosis, disease duration, ever smoking, extensive disease (pancolitis), the first 3 PCs (for population stratification), and the identified 3 variants (rs3131621, rs9275596, and rs11244).
SNP rs3131621, located at 6p21.33, is a synonymous variant between HLA-B and MICA/MICB (Figure 1a). In the discovery cohort, minor and risk allele G was associated with an increased PSC risk (allelic OR, 2.60; P = 1.44E-05). In the replicate cohort, allele G was associated with an increased PSC risk (allelic OR, 3.81; P = 0.001). In the combined cohort, allele G was associated with an increased PSC risk (allelic OR, 2.81; P = 1.03E-07) (Table 2). In the multivariate logistic regression model with additive genetic mode, rs3131621 remained significantly associated with PSC risk (adjusted OR, 2.00; P = 0.002) (Table 2).
 Figure 1.:
Figure 1.: Genetic variants associated with risk of PSC in patients with UC. (a), rs3131621 (GRCh38 chr06: 31,457,722) located at 6p21.32 with nearby genes HLA-B, MICA/MICB, and LTA (6:31,325,000–31,580,000). (b) rs9275596 (GRCh38: chr06: 32,713,854) and rs11244 (GRCh38: chr06: 32,812,947) located at 6p21.33 with nearby genes HLA-DQB1, HLA-DQA2, HLA-DQB2, HLA-DOB, and PSMB8/9 (6:32,600,000–32,900,000). GRCh38, Genome Reference Consortium Human Build 38; IBD, inflammatory bowel disease; LD, linkage disequilibrium; MAF, minor allele frequency; mb, megabase; NCBI, National Center for Biotechnology Information; PSC, primary sclerosing cholangitis; RefSeq, reference sequence; SNPs, single-nucleotide polymorphisms; UC, ulcerative colitis.
The other 2 SNPs (rs9275596 and rs11244) were found located at 6p21.32. SNP rs9275596 is a synonymous variant between HLA-DQB1, HLA-DQA2, and HLA-DQB2 (Figure 1b). In the discovery cohort, minor and risk allele C was associated with an increased PSC risk (allelic OR, 3.31; P = 1.15E-07). In the replicate cohort, allele C was associated with an increased PSC risk (allelic OR, 3.03; P = 0.009). In the combined cohort, allele C was associated with an increased PSC risk (allelic OR, 3.36; P = 8.75E-10) (Table 2). In the multivariate logistic regression model with additive genetic mode, rs9275596 remained significantly associated with PSC risk (adjusted OR, 2.17; P = 0.001) (Table 2).
SNP rs11244 is a synonymous variant at 3' untranslated region of HLA-DOB and nearby TAP1/2 and PSMB8/9 (Figure 1b). In the discovery cohort, minor and risk allele A was associated with an increased PSC risk (allelic OR, 3.36; P = 2.84E-08). In the replicate cohort, allele A was associated with an increased PSC risk (allelic OR, 3.04; P = 0.008). In the combined cohort, allele A was associated with an increased PSC risk (allelic OR, 3.26; P = 8.64E-10) (Table 2). In the multivariate logistic regression model with additive genetic mode, rs11244 remained significantly associated with PSC risk (adjusted OR, 2.29; P = 0.0005) (Table 2).
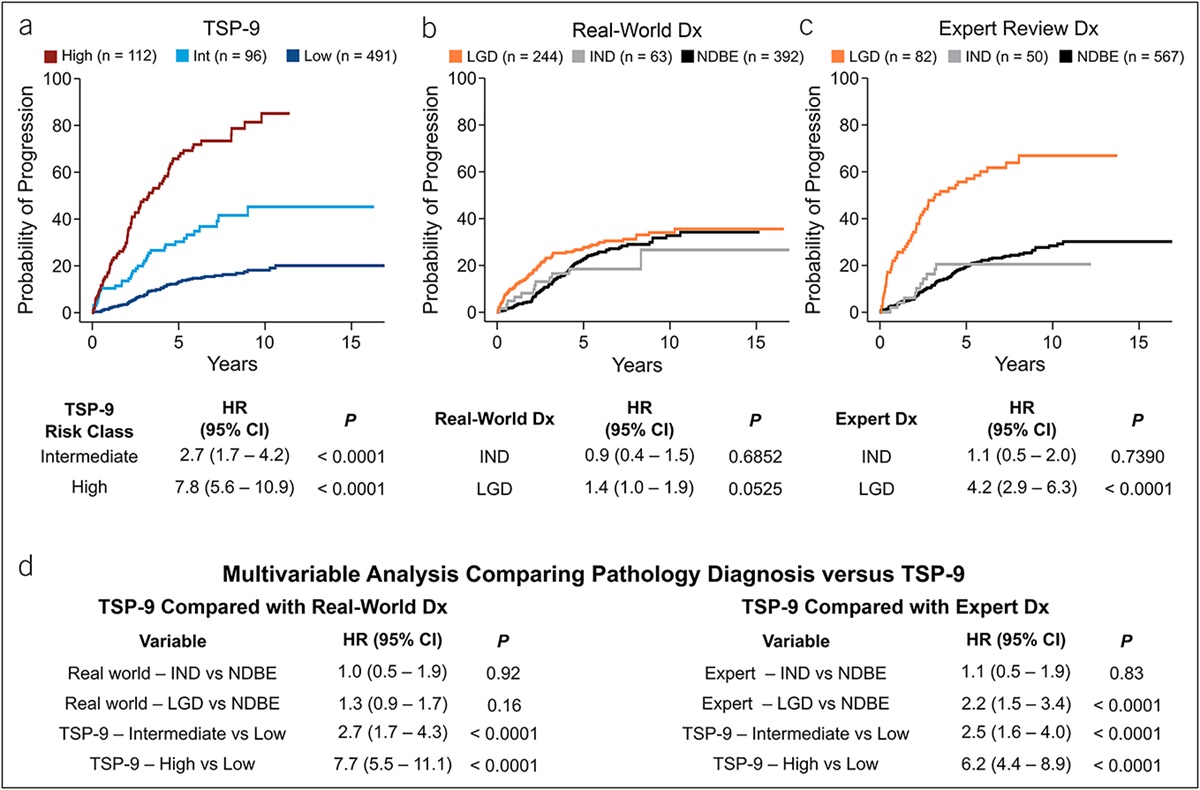
Model predictabilityUsing AUC, we assessed model discriminatory power of clinical predictors (age at UC diagnosis, disease duration, ever smoking, and extent of disease) and combined clinical and genetic predictors (rs3131621, rs9275596, rs11244 in additive genetic mode) in the combined cohort. For clinical predictors, AUC increased from 82% to 88% after adding the genetic predictors (P < 0.001); explained PSC in UC variance, estimated by McFadden pseudo R2, increased from 20% to 33% (Figure 2).
 Figure 2.:
Figure 2.: Receiver operating characteristic curves comparing model predictability. Left, clinical predictors only (age at UC diagnosis, disease duration, ever smoking, and extent of disease). Right, combined clinical and genetic predictors (rs3131621, rs9275596, rs11244 in additive mode). Likelihood ratio test showed P < 0.001. AUC, area under the curve; SNPs, single-nucleotide polymorphisms.
DISCUSSIONThrough a GWAS search in a long-term follow-up IBD GWAS discovery cohort replicated by another independent IBD cohort, we identified 3 genetic variants: rs3131621, located at 6p21.33 (between HLA-B, MICA/MICB, and LTA) and rs9275596 and rs11244, both located at 6p21.32 (between HLA-DQB1 and HLA-DQA2/B2 [rs9275596] and HLA-DOB, TAP1/2, and PSMB8/9 [rs11244]). These variants were found to distinguish patients with UC-PSC from those with UC alone. The identified signal at 6p21.32 (HLA region) in this study was also reported in findings from a recent PSC GWAS meta-analysis (13). Compared with patients with UC alone, patients with UC-PSC were younger when diagnosed with UC and had lower rates of smoking and more extensive disease. We established a model integrating both clinical and genetic predictors capable of predicting PSC risk in patients with UC (AUC, 88%).
The HLA complex—stretching across 7.6 million base pairs of DNA on the short arm of chromosome 6—contains 252 genes, 28% of which are related to immunologic response (17). Due to the high LD between alleles at the HLA class I, II, and III loci, an extended HLA haplotype with HLA-B8 alleles (ie, HLA-B*08) and HLA-DR3 (ie, DRB1*0301) has been found remarkably conserved in those with Northern European ancestry. This haplotype is associated with a wide range of autoimmune diseases, including PSC (18,19). The genetic association between variants in HLA and PSC was first reported in 1982 (20,21). Later studies verified that PSC-associated loci also exist for the other alleles at this extended high-LD region (eg, HLA-A1, HLA-C7, MICA*008/5.1, and the tumor necrosis factor α promoter -308 A alleles) (22–25). The most prominent PSC-associated variants include the risk haplotypes of HLA-A*01-C*07-B*08-DRB1*0301-DQB1*0201 and DRB1*1301-DQB1*0603 and the protective allele DRB1*04 (26,27), which was also found to have a protective effect in UC (28). In both UC and PSC, disease associations have been demonstrated for several genetic variants within the HLA complex on chromosome 6p21 (13,29–31).
In a recent analysis estimating the genetic correlation (rG) between PSC and subphenotypes of IBD by the National Institute of Diabetes and Digestive and Kidney Diseases Inflammatory Bowel Disease Genetics ConsortiuM (31), it was found that PSC is more genetically related to UC (rG = 0.29) than CD (rG = 0.04, P = 2.55E−15) (9), but the rG is still lower than that between UC and CD (rG = 0.56). These findings suggest that from a genetic perspective, UC-PSC may not be the same as UC alone and that other predictors (eg, shared environmental factors) may also play a role.
It is of interest for clinicians to know whether genetic associations detected in PSC may help distinguish patients with UC-PSC from those with UC alone. In an earlier study, HLA-B8 was found more frequently in patients with UC-PSC than those with UC alone (RR, 8.36; P < 0.001), as was the case in HLA-DR3 (RR, 4.28; P < 0.025) (21). In another study of HLA in patients with UC-PSC (32), no association was found among the main PSC risk alleles (DRB1*0301, DRB1*1301, DRB1*1501) in patients with UC. In our study, 3 genetic variants (rs3131621 at 6p21.33 and rs9275596 and rs11244, both at 6p21.32) were found to distinguish patients with UC-PSC from those with UC alone. These signals correspond to the HLA region on chromosome 6p21.3, which was previously found associated with PSC.
In particular, the signal of rs9275596 (at 6p21.32: GRCh38 chr06: 32,713,854) in our study was physically located next to the PSC risk variant rs9275171 (at 6p21.32: GRCh38 chr06: 32,686,195) found in a recent PSC GWAS meta-analysis (13). SNP rs3131621, a synonymous variant between HLA-B and MICA/MICB (Figure 1a), and risk allele G were associated with a 2-fold increased PSC risk. SNP rs9275596, between HLA-DQB1, HLA-DQA2, and HLA-DQB2 (Figure 1b), and risk allele C were associated with a 3-fold increased risk of PSC. SNP rs11244, synonymous variant at 3' untranslated region of HLA-DOB and nearby TAP1/2 and PSMB8/9 (Figure 1b), and risk allele A were associated with a 3-fold increased risk of PSC.
Several clinical features (mild disease course, rectal sparing, high frequency of pancolitis, predominant inflammation following a proximal-to-distal colon distribution, and backwash ileitis) observed in patients with UC-PSC may indicate that UC-PSC is a distinct IBD subphenotype (6). In our study, compared with patients with UC alone, patients with UC-PSC were more likely to be younger at age of diagnosis and had longer disease duration, less smoking history, and more extensive disease. This further supports that patients with UC-PSC compose a unique subgroup of patients with UC.
Some limitations of our study warrant consideration. First, this was a retrospective study, and the number of patients with UC-PSC was relatively small. Second, the exact date of PSC diagnosis was not always available, and we were unable to determine the time interval between UC diagnosis and PSC diagnosis. Third, the IBD GWAS cohorts consisted of only White patients, thus limiting generalizability.
Our study had several strengths. We had multiple independent, large, long-term follow-up IBD cohorts (serving as discovery and replicate datasets), and we focused on clinically relevant characteristics to identify predictors of PSC risk in patients with UC. In addition, the included studies met quality control criteria, according to the methodologic quality assessment in IBD clinical behavior and phenotyping and genotyping data.
It is clinically important to identify the high-risk subgroup of patients with UC who will likely develop PSC because once diagnosed it may progress to liver failure within 1–2 decades. There is also an increased risk of cancer among patients with UC-PSC; for example, cholangiocarcinoma and other gastrointestinal malignancies (i.e., pancreatic and colorectal cancer). Through our constructed discriminatory UC-PSC model, a patient with UC who never smoked, was diagnosed at a younger age (<20 years), was with extended disease, and was carrying all 3 risk variants (rs3131621, rs9275596, rs11244) would have a much higher risk of PSC than a patient with UC who smoked, was diagnosed at an older age, was without extended disease, and was not carrying any of the 3 risk variants. For those patients with high-risk UC, screening for PSC (e.g., magnetic resonance cholangiopancreatography) may be considered.
In conclusion, we successfully identified 3 genetic variants (rs3131621, rs9275596, and rs11244) that significantly distinguish patients with UC-PSC from those with UC alone. The identified signals of the HLA region at 6p21.32 correlate with findings of PSC loci through a recent GWAS meta-analysis. Our predictive model integrating clinical and genetic predictors has the potential to identify patients with UC who may also have or will develop PSC and, used as a personalized medicine-based tool, could help guide different clinical management and monitoring strategies.
CONFLICTS OF INTERESTGuarantor of the article: Ming-Hsi Wang, MD, PhD.
Specific author contributions: M.-H.W.: contributed to the conception and design, analysis and interpretation of the data, drafting and critical revision of the article, and generation/collection of figures. J.J.F.: contributed to the experiments and collection of data. N.R.: contributed to the experiments and collection of data. K.M.: contributed to the experiments and collection of data. B.D.N.: contributed to the experiments and collection of data. C.F.: contributed to the conception and design and critical revision of the article. L.E.R.: contributed to the conception and design and critical revision of the article. J.A.L.: contributed to the conception and design and critical revision of the article. S.F.P.: contributed to the conception and design and critical revision of the article. M.F.P.: contributed to the conception and design and critical revision of the article. R.D.N.: contributed to the study cohort establishment, conception and design, and critical revision of the article. J.P.A.: contributed to the study cohort establishment, conception and design, and critical revision of the article. W.A.F.: contributed to the study cohort establishment, conception and design, and critical revision of the article.
Financial support: This study was supported by the Helmsley Charitable Trust and Mayo Clinic Health System Intramural Grant. The funding bodies had no role in the study design, the collection, analysis, and interpretation of data, or the writing of the article and the decision to submit it for publication.
Potential competing interests: None to report.
IRB approval statement: All data collection and study procedures were approved by the institutional review board of each cohort's respective institution (Mayo Clinic IRB, Cleveland Clinic IRB, Washington University at St Louis IRB).
Study Highlights
WHAT IS KNOWN ✓ Primary sclerosing cholangitis (PSC) uniquely influences disease behavior and outcomes in patients with ulcerative colitis (UC); however, early detection of PSC through a set of integrated clinical and genetic predictors remains to be defined. WHAT IS NEW HERE ✓ Through a collaborative multitertiary centers' hypothesis-free genetic association approach, for the first time, we identified novel genetic variants, of genome-wide significance, which distinguished patients with UC-PSC from patients with UC alone. ✓ Our predictive model integrating clinical and genetic predictors has the potential to identify patients with UC who may also have or will develop PSC and, used as a personalized medicine-based tool, could help guide different clinical management and monitoring strategies. ACKNOWLEDGMENTThe Scientific Publications staff at Mayo Clinic provided copyediting support.
REFERENCES 1. Boonstra K, van Erpecum KJ, van Nieuwkerk KMJ, et al. Primary sclerosing cholangitis is associated with a distinct phenotype of inflammatory bowel disease. Inflamm Bowel Dis 2012;18:2270–6. 2. Ponsioen CY, Vrouenraets SM, Prawirodirdjo W, et al. Natural history of primary sclerosing cholangitis and prognostic value of cholangiography in a Dutch population. Gut 2002;51:562–6. 3. Loftus EV Jr., Harewood GC, Loftus CG, et al. PSC-IBD: A unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut 2005;54:91–6. 4. Sano H, Nakazawa T, Ando T, et al. Clinical characteristics of inflammatory bowel disease associated with primary sclerosing cholangitis. J Hepatobiliary Pancreat Sci 2011;18:154–61. 5. Soetikno RM, Lin OS, Heidenreich PA, et al. Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis: A meta-analysis. Gastrointest Endosc 2002;56:48–54. 6. de Vries AB, Janse M, Blokzijl H, et al. Distinctive inflammatory bowel disease phenotype in primary sclerosing cholangitis. World J Gastroenterol 2015;21:1956–71. 7. Bergquist A, Lindberg G, Saarinen S, et al. Increased prevalence of primary sclerosing cholangitis among first-degree relatives. J Hepatol 2005;42:252–6. 8. Mathew CG, Lewis CM. Genetics of inflammatory bowel disease: Progress and prospects. Hum Mol Genet 200413:R161–168. 9. Ji SG, Juran BD, Mucha S, et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet 2017;49:269–73. 10. Ellinghaus D, Jostins L, Spain SL, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet 2016;48:510–8. 11. Karlsen TH, Franke A, Melum E, et al. Genome-wide association analysis in primary sclerosing cholangitis. Gastroenterology 2010;138:1102–11. 12. Liu JZ, Hov JR, Folseraas T, et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet 2013;45:670–5. 13. Cordell HJ, Fryett JJ, Ueno K, et al. An international genome-wide meta-analysis of primary biliary cholangitis: Novel risk loci and candidate drugs. J Hepatol 2021;75:572–81. 14. Ellinghaus D, Folseraas T, Holm K, et al. Genome-wide association analysis in primary sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF4. Hepatology 2013;58:1074–83. 15. Dassopoulos T, Nguyen GC, Bitton A, et al. Assessment of reliability and validity of IBD phenotyping within the national institutes of diabetes and digestive and kidney diseases (NIDDK) IBD genetics consortium (IBDGC). Inflamm Bowel Dis 2007;13:975–83. 16. McFadden D. Conditional logit analysis of qualitative choice behavior. In: Zaremba P (ed). Frontiers in Econometrics. Academic Press, Econometrics Laboratory: UC Berkeley, 1974, pp 105–42. 17. Horton R, Wilming L, Rand V, et al. Gene map of the extended human MHC. Nat Rev Genet 2004;5:889–99. 18. Candore G, Lio D, Colonna Romano G, et al. Pathogenesis of autoimmune diseases associated with 8.1 ancestral haplotype: Effect of multiple gene interactions. Autoimmun Rev 2002;1:29–35. 19. Price P, Witt C, Allcock R, et al. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol Rev 1999;167:257–74. 20. Chapman RW, Varghese Z, Gaul R, et al. Association of primary sclerosing cholangitis with HLA-B8. Gut 1983;24:38–41. 21. Schrumpf E, Fausa O, Forre O, et al. HLA antigens and immunoregulatory T cells in ulcerative colitis associated with hepatobiliary disease. Scand J Gastroenterol 1982;17:187–91. 22. Donaldson PT, Farrant JM, Wilkinson ML, et al. Dual association of HLA DR2 and DR3 with primary sclerosing cholangitis. Hepatology 1991;13:129–33. 23. Moloney MM, Thomson LJ, Strettell MJ, et al. Human leukocyte antigen-C genes and susceptibility to primary sclerosing cholangitis. Hepatology 1998;28:660–2. 24. Neri TM, Cavestro GM, Seghini P, et al. Novel association of HLA-haplotypes with primary sclerosing cholangitis (PSC) in a southern European population. Dig Liver Dis 2003;35:571–6. 25. Norris S, Kondeatis E, Collins R, et al. Mapping MHC-encoded susceptibility and resistance in primary sclerosing cholangitis: The role of MICA polymorphism. Gastroenterology 2001;120:1475–82. 26. Donaldson PT, Norris S. Evaluation of the role of MHC class II alleles, haplotypes and selected amino acid sequences in primary sclerosing cholangitis. Autoimmunity 2002;35:555–64. 27. Spurkland A, Saarinen S, Boberg KM, et al. HLA class II haplotypes in primary sclerosing cholangitis patients from five European populations. Tissue Antigens 1999;53:459–69. 28. Stokkers PC, Reitsma PH, Tytgat GN, et al. HLA-DR and -dq phenotypes in inflammatory bowel disease: A meta-analysis. Gut 1999;45:395–401. 29. Donaldson PT, Norris S. Immunogenetics in PSC. Best Pract Res Clin Gastroenterol 2001;15:611–27. 30. Yap LM, Ahmad T, Jewell DP. The contribution of HLA genes to IBD susceptibility and phenotype. Best Pract Res Clin Gastroenterol 2004;18:577–96. 31. Liu JZ, van Sommeren S, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015;47:979–86. 32. Karlsen TH, Boberg KM, Vatn M, et al. Different HLA class II associations in ulcerative colitis patients with and without primary sclerosing cholangitis. Genes Immun 2007;8:275–8.
Comments (0)