
Remember me
Hirschsprung disease (HSCR) is a life-threatening disorder in children, with a worldwide incidence of 1 in 5,000 live births. It is caused by aganglionosis in the distal bowel resulting in a spastic segment of the colon, which does not allow adequate fecal evacuation. Left untreated, affected children have significant morbidity related to malabsorption, anemia, failure to thrive, enterocolitis, intestinal obstruction, and toxic megacolon. Though considered a disease of multigenic inheritance, complex gene-environmental interactions have been shown to play an important role in its etiology. Vitamin A deficiency and its interaction with the REarranged during Transfection (RET) proto-oncogene is one such risk factor of HSCR identified in rodents (1).
The RET proto-oncogene is a neural crest cell migration regulator that is relevant in the pathogenesis of HSCR (2). The active form of vitamin A, retinoic acid, regulates RET transcription by binding to retinoic acid response elements in its promoter region. Perturbation in the retinoic acid pathway during early embryogenesis has been shown, in animal studies, to consistently interfere with key events of normal bowel innervation, such as enteric nervous system precursor proliferation, differentiation, and migration (3–7). There are, however, no human studies that have demonstrated this association.
We hypothesized that in humans, a low maternal vitamin A status in early embryogenesis (first trimester) may be linked to the development of HSCR in the fetus and that this low vitamin A status potentiates RET gene–related risk of HSCR. To prove this in humans, 2 difficulties arise: the accurate measurement of vitamin A status and its timing. The most accurate method to measure vitamin A status is the measurement of its body (liver) stores by a stable isotope dilution approach. Unlike serum retinol, this technique is sensitive and accurate and has been successfully used to assess the efficacy of vitamin A interventions (8).
The earliest that HSCR can be diagnosed is in the first few days of postnatal life, whereas the role of vitamin A in the etiopathogenesis of the disease is in the early antenatal period. However, early postnatal maternal evaluations, soon after HSCR diagnosis, offers the possibility of evaluating risk in a case-control design. Therefore, postnatal measurements of the pool size in mothers of children with HSCR may be a reliable surrogate of their early antenatal vitamin A status, if there were no drastic changes or supplementation of vitamin A intake during pregnancy. Vitamin A status has also been previously reported to be relatively stable across the postnatal period (0–20 months) (9). This study therefore aimed to evaluate and compare the early postnatal maternal vitamin A pool size in mothers of children with HSCR with those in mothers of normal children. Furthermore, pathogenic variants in RET exons and its promoter region were investigated in children with HSCR and their parents. Untargeted maternal plasma metabolomic profiling was also performed to evaluate potential metabolite patterns reflecting risk.
METHODS Study populationThis observational case-control study was conducted at St. John's Medical College Hospital, Bengaluru, India, over a period of 8 months from June 2019 to January 2020 after obtaining institutional ethics approval (IEC-89/2019). The vitamin A pool size by modified relative dose response in postpartum women has been reported to be 0.0480 ± 0.037 (10). To detect a difference of 70% in modified relative dose response between groups with standard deviating half of the mean, at 80% power and 5% level of significance, a sample size of 7 mothers of children with HSCR and 7 control mothers of children without HSCR was determined. Consenting mothers (n = 7) of infants aged 1 year or younger diagnosed with HSCR were recruited after diagnosis by histopathological biopsy of the colon or rectum, with or without radiological evidence by contrast enema. Consenting mothers (n = 6, controls) of children with no developmental or biochemical anomalies, matched for their age and the age of their child, were also recruited. Mothers with excess gestational weight gain (>20 kg) or weight loss (>5 kg), those with clinical signs of vitamin A deficiency, or those on regular medications such as statins, antidepressants, and antipsychotics or on vitamin A supplements were excluded. Dietary recall of foods consumed in the preconceptual period was collected by food frequency questionnaire to assess pattern of intake of vitamin A rich foods in both cases and controls.
Measurement of body vitamin A storesThe measurement of maternal vitamin A stores was performed by administering a single oral dose of tetra-deuterated retinyl acetate (2.5 mg of [2H4]-retinyl acetate; Cambridge Isotope Laboratory, MA). The dose, prepared in corn oil, was administered along with a small amount of bread (16 g) and butter (1.7 g) on day 0 in the fasted state, after the collection of a baseline blood sample. Additional blood samples were also collected on day 1 (24 hours), day 3, and day 21 after dosing. Venous blood samples, drawn into ethylenediaminetetraacetic acid vacutainers (BD Vacutainer, Mumbai, India) covered with aluminium foil to prevent light exposure, were centrifuged for 15 minutes at 3,500 rpm at 4 °C for separation of plasma, which was aliquoted into cryovials and stored at −80 °C.
Plasma samples were purified by solid-phase extraction as described in the Supplementary Digital Content (see Supplementary Methods, https://links.lww.com/CTG/A979) and derivatized using a derivatization mix of 4:1:1 (Decane:Pyridine:1-[tert-butyldimethylsilyl] imidazole). These samples were analyzed by gas chromatography (GC)-mass spectrometry in selected ion monitoring mode. GC separation was performed using a DB-5MS capillary column (Agilent J&W). The GC oven temperature was maintained at 190 °C for 1 min, increased to 270 °C at a rate of 4 °C/min, and then to 300 °C at a rate of 32 °C/min and then maintained for 1 minute. The injector temperature was set to 285 °C. The isotopic ratio of 2H4-retinol to unlabeled retinol was determined by monitoring fragment ions at mz 401 and 404 for retinol and [2H4]-retinol, respectively. The isotope ratio assay precision was estimated periodically by analyzing known standards with each set of plasma samples, with an intraday and interday coefficient of variation of 3 and 5%, respectively.
The total body vitamin A pool size (mmol retinol) was quantified using the measured plasma isotopic dilution of labeled retinol on day 3, using the Green equation (11). The pool size obtained by this stable isotope dilution method was converted to liver vitamin A concentrations by assuming that liver weight was 2.4% of body weight and that the liver contained 90% of total body vitamin A (12).
Targeted amplicon next-generation sequencing of RET exons and a 3.5-kb RET upstream regionGenomic DNA from buffy coat of blood was extracted using Qiagen DNeasy Blood and tissue Kit (QIAGEN Inc, MD, Cat#69506), as described in Supplementary Digital Content (see Supplementary Methods, https://links.lww.com/CTG/A979). RET exons and a 3.5-kb upstream region (13) were polymerase chain reaction amplified (primers sequences are available in Supplementary Digital Content [see Supplementary Table 2, https://links.lww.com/CTG/A979]). The amplicons from individual samples were pooled in equimolar concentration and purified using Sera-MagTM Select beads (Cytiva, Buckinghamshire, UK, Cat#29343057), quantified using Qubit fluorometer (Thermo Fisher Scientific, MA), and further subjected to Illumina library preparation, using QIASeq FX DNA Library Preparation protocol (QIAGEN, MD, Cat#180475) by following manufacturer's instructions. Library preparation details are available in Supplementary Digital Content (see Supplementary Methods, https://links.lww.com/CTG/A979). The fragment size distribution of the final sequencing library pools was analyzed on Agilent TapeStation.
Data obtained from the sequencing run was processed as detailed in Supplementary Digital Content (see Supplementary Methods, https://links.lww.com/CTG/A979). The amplicon sequencing analyses were performed at Genotypic Technology Pvt. Ltd. Bangalore, India.
Next generation sequencing data analysis of RET exons and 3.5 kb upstream regionAdapter-clipped, high-quality reads were aligned against human reference genome (GRCh38.p14) (14) using BWA-v0.7.5 tool (15). The processing of the alignment data was performed using Samtools-v1.94 (16), and polymerase chain reaction duplicate reads were removed using PICARD-v1.1025 (17). For identification of variants, 2 different tools, –GATK-v4.1.4.1 (18) HaplotypeCaller and Bcftools-1.124 (16), were used. Similarly, joint variant calling was performed for each proband-parent trio using GATK-v4.1.4.16 GenotypeGVCFs and Bcftools-1.124 to identify germline mutations. The functional annotation of the variants were performed using wAnnovar (19). Low-quality variant calls with coverage depth lower than 100 were excluded. Variants of potential clinical significance were identified by excluding (i) silent single-nucleotide variants, nonframeshift insertions/deletions, and intronic variants and (ii) variants occurring with a minor allele frequency > 5% in the IndiGen (20) (Indian population-specific database), 1000 Genomes (21) (all populations), or GnomAD (22) (all populations) databases. The pathogenicity of the missense variants was predicted using VarSome (23) tool. The expression quantitative trait loci for RET gene in colon-sigmoid and colon-transverse tissues were obtained from the Genotype-Tissue Expression portal (https://gtexportal.org). Variant phasing for the haplotype analysis was performed with South Asian population from the 1000 Genome project (phase 3) as the reference panel using SHAPEIT v4.1.3 (24).
STATISTICSNormally distributed data were reported as mean and SD, while skewed data were reported as median (Q1, Q3). The significance of differences between skewed data was determined using the Mann-Whitney U test. Analyses were performed using SPSS, and P values were considered statistically significant at the 5% level (P < 0.05).
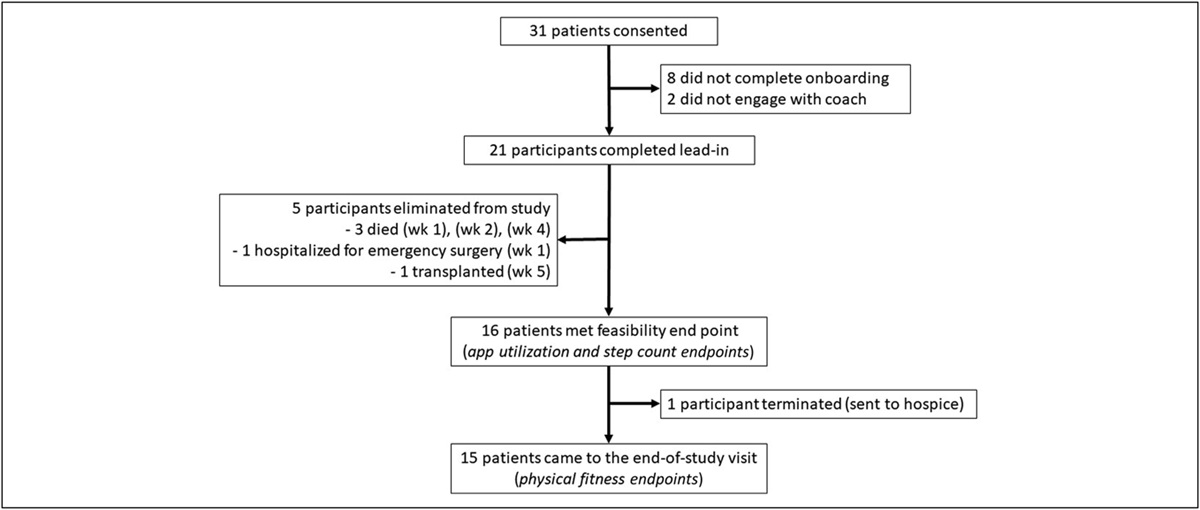
RESULTSA total of 13 subjects were recruited and completed the 3-day blood sampling protocol after tracer dosing for measurement of body vitamin A stores (Figure 1). Demographic, anthropometric, and dietary information of subjects of children with (cases) and without (controls) HSCR are summarized in Table 1. The groups were comparable in these characteristics at recruitment. The median time of recruitment of subjects was within the first month after delivery (see Supplementary Table 1, https://links.lww.com/CTG/A979). The average birth weight of children with HSCR was 3.01 ± 0.45 kg and that of the normal children was 2.78 ± 0.82 kg. The mean vitamin A body store was 1,158 ± 966 μmol. Although all subjects had liver vitamin A stores that were in the normal range (0.2–1.6 μmol/g calculated liver tissue), the mothers of children with HSCR had vitamin A stores that were nearly half (0.50 ± 0.17 μmol/g, n = 7) that of those in the controls, tending toward significance (0.89 ± 0.51 μmol/g, n = 6, P = 0.079, Table 2).
 Figure 1.:
Figure 1.: Consort diagram.
Table 1. - Demographics, anthropometry, dietary information, and laboratory analysis parameters between mothers of children with HSCR and those of control children Maternal characteristics HSCR (n = 7) Control (n = 6) P value Age, y 25.1 ± 2.5 28.3 ± 5.9 0.22 Anthropometry Weight, kg 55.3 ± 11.1 64.7 ± 17.3 0.26 Height, cm 154.5 ± 4.5 159.6 ± 5.4 0.09 BMI, kg/m2 23.1 ± 3.8 25.6 ± 7.1 0.43 Diet Energy, kcal/d 1,780.1 ± 249.5 1,809.5 ± 334.1 0.86 Carbohydrate, g/d 236.6 ± 68.9 231.4 ± 40.5 0.87 Protein, g/d 51.7 ± 20.7 69.6 ± 25.2 0.52 Fat, g/d 69.6 ± 25.2 72.3 ± 23.0 0.85 Vitamin A, μg RAE/d 296.6 ± 130.3 326.0 ± 237.8 0.78Mean ± SD.
BMI, body mass index; HSCR, Hirschsprung disease.
Mean ± SD.
HSCR, Hirschsprung disease.
From targeted amplicon sequencing of the RET exons and a 3.5-kb upstream region (13), an average of 1.6 million processed reads per sample were obtained (see Supplementary Table 2, https://links.lww.com/CTG/A979). Two novel de novo mutations were identified in 2 cases (Table 3). A single-nucleotide deletion, rs369660849 (T>del), was detected at Chr10: 43074338 in the 3.5-kb RET upstream region in all 7 cases and in a heterozygous state in both parents (Table 4). Haplotype A-G-G involving allele A of rs1800858 (c.A135G, p.A45A), allele G of rs1800860 (c.A1296G, p.A432A), and allele G of rs1800861 (c.G2307T, p.L769L) were detected in 5 of 7 cases (Table 5). Five common noncoding RET variants (rs12267460, rs2435351, rs2505530, rs2472739, and rs2435352) that are known regulators (expression quantitative trait locis) of RET expression (in colon-sigmoid and colon-transverse tissues) were identified in the cases and parents of 3 trios (Table 6-VAHDCA_03C, VAHDCA_04C, and VAHDCA_05C).
Table 3. - RET germline mutations identified in 7 cases and their unaffected parents Position Variant ID HGVS Type Location Exon AA change Annotation Ref Alt Patients Unaffected family Reports CADD Phylop100Way IndiGen AF 1KGP3-ALL AF gnomAD genomes AF chr10:43123726 — NM_020975:c.2857delC Deletion Exonic 17 p.P953Lfs*12 Frameshift C — VAHDCA_04C (hom) — Novel 33 7.84 — — — chr10:43118409 — NM_020975:c.2321C>G SNV Exonic 13 p.S774X Stop gain C G VAHDCA_05C (hom) — Novel 43 7.76 — — — chr10:43124919 rs528823385 NM_020975:c.2976G>A SNV Exonic 18 p.P992P Synonymous G A VAHDCA_03C (het), VAHDCA_05C (het) VAHDCA_03B (het), VAHDCA_05B (het) Clinvar benign — — 0.0175 0.0052 0.0000323 chr10:43128257 rs753203348 NM_020975:c.3333G>A SNV Exonic 20 p.T1111T Synonymous G A VAHDCA_05C (het) VAHDCA_05A (het) Reported likely benign for other conditions — — — — 1.40E-05 chr10:43074338 rs369660849 -2731 upstream of RET TSS Deletion Intergenic — — — T — VAHDCA_01C (het), VAHDCA_02C (het), VAHDCA_03C (het), VAHDCA_04C (het), VAHDCA_05C (het), VAHDCA_06C (het), VAHDCA_07C (het) VAHDCA_01A (het), VAHDCA_01B (het), VAHDCA_02A (het), VAHDCA_02B (het), VAHDCA_03A (het), VAHDCA_03B (het), VAHDCA_04A (het), VAHDCA_04B (het), VAHDCA_05A (het), VAHDCA_05B (het), VAHDCA_06A (het), VAHDCA_06B (het), VAHDCA_07A (het), VAHDCA_07B (het) Not reported — — — — 0.0227 chr10:43074350 rs370474445 -2719 upstream of RET TSS SNV Intergenic — — — T C VAHDCA_02C (het) VAHDCA_02B (het) Not reported — — 0.0362 0.011 0.0001 chr10:43128845 rs185408658 NM_020975:c.*576G>A SNV UTR3 — — — G A VAHDCA_03C (het) VAHDCA_03A (het) Clinvar likely benign — — 0.0049 0.0022 0.0002AA, amino acid; AF, allele frequency; CADD, combined annotation–dependent depletion; HGVS, Human Genome Variation Society; RET, REarranged during Transfection; SNP, single nucleotide variation; TSS, transcription start site; 1KGP3-ALL, 1000 Genomes Project full panel (phase 3).
1KGP3: 1000 Genomes Project (phase 3); SAS, South Asian population.
Cases: VAHDCA_01–07; Mothers: VAHDCA_01–07A; Fathers: VAHDCA_01-7B.
RET, REarranged during Transfection.
eQTL, expression quantitative trait loci; RET, REarranged during Transfection.
1KGP3: 1000 Genomes Project (phase 3).
This study found that the postpartum vitamin A liver store in mothers of children born with HSCR was lower (by approximately half) than that of mothers of normal children though the liver stores in both groups were within normal range (0.2–1.6 μmol/g of liver weight). The stable isotope method used in this study for measuring liver stores is the current gold standard of measuring vitamin A status in a human subject, and the 3-day protocol that was used to reduce participant burden has been validated in previous studies in field and clinical experiments (25).
Retinol binding protein–deficient (Rbp4−/−) mice, which are dependent on dietary retinol sources because they cannot mobilize their hepatic retinoid stores (retinol, retinoic acid, and retinyl ester), have 40% lower fetal retinoid concentrations even when fed a vitamin A–sufficient diet (26). When fed a retinol-deficient diet, pregnant Rbp4−/− mice had a further quantitative reduction of retinoids by 42%–50% (6), and this resulted in an HSCR phenotype in 31 of 33 offsprings compared with 2 of 34 mice fed a retinol-sufficient diet. This reduction of retinoid concentration as an exposure that increases the risk of HSCR is similar to our study, where there was a reduction of 56%–62% of the vitamin A store (per gram liver tissue) in mothers with children with HSCR compared with that in mothers with normal children. It is worth noting that even a 40% reduction in fetal retinoid concentration was sufficient to cause the HSCR phenotype in 94% of offsprings, when Rbp4−/− mice, which were also RET heterozygous (Ret−/+), were used. This implies that in the background of genetic predisposition, even small depletions of vitamin A stores, and hence retinoid availability, can result in HSCR.
In the sequencing of RET exons and a 3.5-kb upstream region, 2 novel de novo mutations in 2 of the 7 cases with HSCR were detected, both of which were predicted to cause premature truncation of the RET protein. Both mutations had pathogenic combined annotation–dependent depletion scores (33 and 43, respectively) along with high PhyloP100way scores (7.84 and 7.76, respectively), suggesting highly deleterious mutations in strongly conserved sites. These variants were also absent from any of the large population-scale variation databases such as gnomAD, 1000 Genomes, and the Indian database, IndiGen. Because the de novo mutations detected were in exons 13 and 17, which code for the cytoplasmic tyrosine kinase containing domain of RET, the resultant mutant RET proteins would have retained their ability to bind to activating growth factors in their extracellular domain but lost their activating tyrosine kinase activity, leading to likely sequestration of the activating growth factors and drastic reduction in signaling through RET, even in the heterozygous state. The heterozygous single-nucleotide deletion (T>del) detected in the 3.5-kb RET upstream region of all cases and in each set of the 7 case parents was predicted by the Ensembl genome browser (27) to be within the RELB proto-oncogene and ZNF384 transcription factor–binding site, which is well known for the ability to regulate embryonic and adult neurogenesis, including enteric neurogenesis (28).
RET is also known to harbor several low-penetrance common SNPs that confer HSCR (29,30) risk. A RET haplotype involving allele A of rs1800858 (c.A135G, p.A45A), allele G of rs1800860 (c.A1296G, p.A432A), and allele G of rs1800861 (c.G2307T, p.L769L), which represented 66% of Chinese patients with HSCR (31) vs 37% in controls (P = 0.009), was identified in 5 of 7 cases compared with that in 8 of 14 unaffected parents. The incomplete disease penetrance of this haplotype suggests the possibility of its linkage with an unknown retinoic acid responsive locus that was likely triggered by lower maternal vitamin A stores in the cases. Of interest, of the 5 common noncoding RET variants identified in the cases and parents of 3 trios, the variant rs2435351 is reported to be likely benign by ClinVar (32) with an allele frequency of 26% in the Indian population.
The presence of disease (HSCR) in the absence of established pathogenic RET variants and relatively benign RET haplotypes but in the background of a lower maternal vitamin A store points to a gene-environmental interaction, with lower maternal vitamin A stores acting as the environmental trigger and likely lower RET expression due to the presence of variants as the genetic trigger. When viewed individually, both the environmental and the genetic factors are benign but in synergy can result in HSCR disease.
These findings, of a lower (although still normal) vitamin A store, may be critical to the prevention of HSCR in children, with its surgical burden and lifelong morbidity of fecal incontinence (50%), constipation (30%), need for bowel washes (10%), and episodes of enterocolitis (33%) (33). This is true particularly for children with syndromic or familial HSCR, where the risk of a sibling being affected is as high as 62% (34), compared with 1%–33% in sporadic HSCR. Therefore, ensuring an adequate periconceptional vitamin A status (35) in mothers from high-risk families may be an important factor for the prevention of HSCR in the fetus. This could take the form of advice to eat foods rich in preformed retinol or carotenoids; alternatively, supplementation in the form of a daily vitamin A dose of 10,000 IU/d until 3 months of confirmed gestation may be considered where dietary retinol or carotenoid intake is low (36). Further investigations into the genetic basis of vitamin A status could enable more personalized dietary recommendations in mothers at high risk of vitamin deficiency (37).
The strength of this study lies in the accurate quantification of liver stores in postpartum mothers, but a limitation is using the postnatal maternal liver store as a surrogate for early antenatal vitamin A status. Care was taken to ensure that the mothers were recruited as early as possible, and in 6 of the 7 cases, HSCR was diagnosed within the first 3 months of life. Care was also taken to match the control mothers for the postpartum month of diagnosis of the cases with HSCR. A food frequency questionnaire was also administered to all participants to ensure that there were no drastic changes in vitamin A intake during the periconceptual period and pregnancy. Another strength of this study lies in the integrated sequencing analysis of the RET coding and upstream region from the cases and their parents, which led to identification of 2 novel pathogenic de novo coding variants.
Besides its small sample size, the limitations of this study include lack of genetic data from non-RET genes, which may influence HSCR risk. Other maternal dietary nutrients such as vitamin D, B12, and folate may interact with lipid metabolism and vitamin A pathways in early pregnancy and need to be additionally explored. Increasing the case-control ratio to increase sample size was not feasible in our study because of the subject burden involved in the isotope dilution method. The sequential blood sampling over 3 days makes the recruitment of control subjects difficult, particularly in the early perinatal period when the mother of a normal baby should ideally be discharged to go home.
In conclusion, mothers of children with HSCR have lower but normal liver stores of vitamin A compared with mothers of normal children. In the background of low maternal vitamin A stores, the occurrence of de novo mutations in the RET exon and the presence of a heterozygous RET upstream variant in all children with HSCR suggests a nutritional/environmental-gene interaction between retinoic acid and RET in the etiology of HSCR. These findings suggest that the improvement of periconceptual vitamin A intake in high-risk mothers might be one way to prevent HSCR.
CONFLICTS OF INTERESTGuarantor of article: Anura V. Kurpad, MD, PhD.
Specific author contributions: S.G.H., S.D., A.M.S. and A.V.K.: designed the study; S.G.H. and A.M.S.: recruited subjects of HSCR group and were involved in patient care; S.G.H. and A.T.: recruited subjects of control group and were involved in patient care; S.D.: executed stable isotope dilution study for vitamin A stores, and data analysis; A.M.S. and A.S.: Next generation sequencing sample and data analysis; S.G.H. and A.V.K.: wrote the first draft of the manuscript with primary responsibility for the final content of the manuscript; all authors: critically reviewed and approved the final version of the manuscript.
Financial support: Supported by the Clinical Research Training Program (CRTP) grant from the Wellcome Trust-Department of Biotechnology (DBT) India Alliance to A.V.K. (IA/CRC/19/1/610006). S.G.H. is a Clinical Research Fellow of the CRTP grant. A.M.S. [IA/CPHI/19/1/504593] and A.S. [IA/E/19/1/504945] are independently supported by Wellcome Trust/DBT India Alliance Fellowships.
Pote
Comments (0)