
Remember me

The patients were examined in the muscle center of the Department of Neurology of Kiel University. Diagnostic genetic testing was performed by the Department of Genetics and Applied Genomics of the University of Tübingen (methods described in [8]). Written informed consent was obtained from all patients. Approval for case reports was obtained from the ethics committee of the medical faculty of Kiel University.
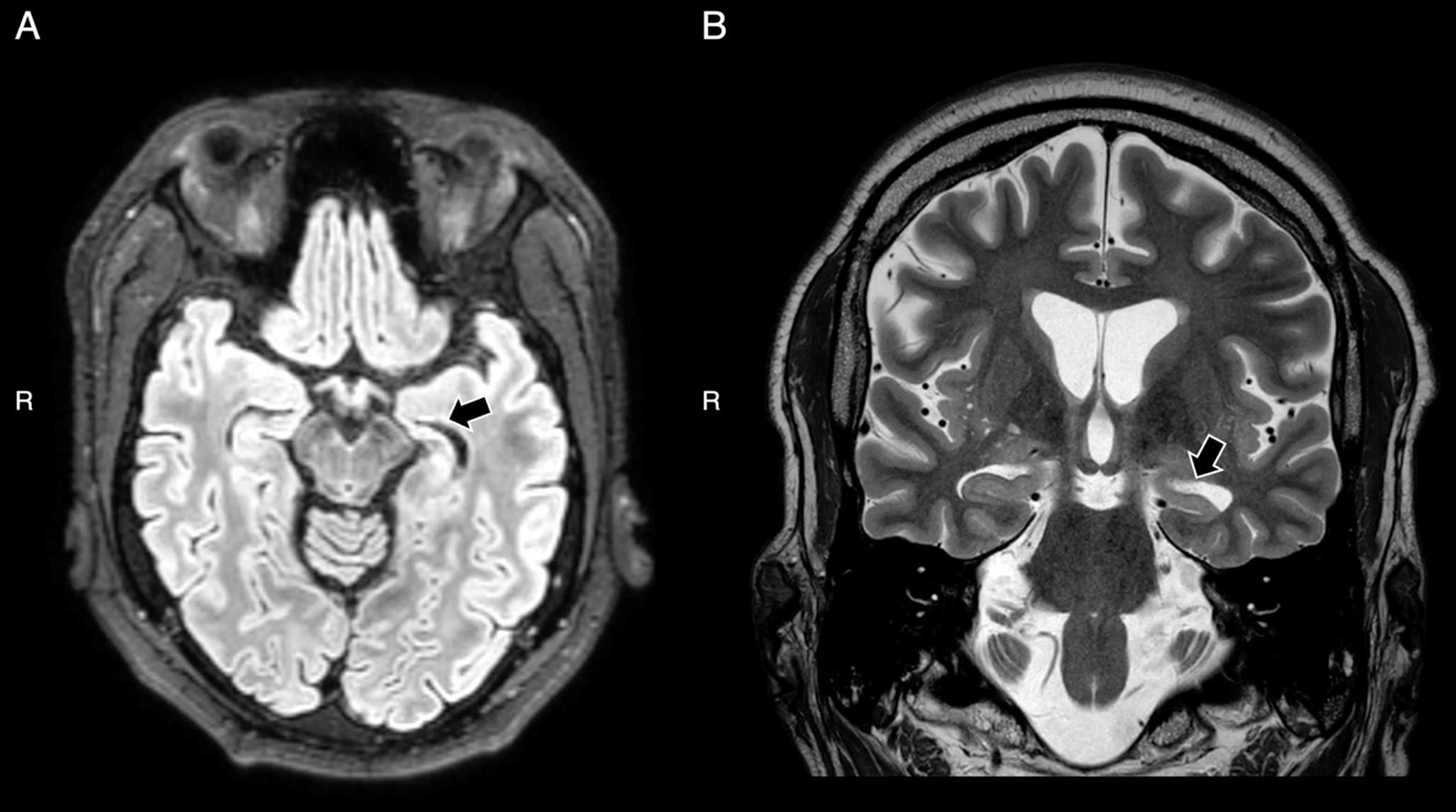
Figure 1 shows the pedigree of the family, Table 1 the clinical characteristics, Table 2 data concerning the identified variants, and supplementary tables 1 and 2 summarize the data obtained from the literature for both variants in question. Five individuals (Fig. 1, c1 to c5) were examined clinically and genetically. All other individuals in generation III are self-reported free of muscular symptoms and did not want predictive genetic diagnostics.
Fig. 1
Pedigree of the family with LGMD calpain 3-related. Empty symbols—clinically unaffected individuals; symbols with black filling—clinically affected individuals carrying both, the NM_000070.3: c.700G>A, p.Gly234Arg and the NM_000070.3: c.1746-20C>G, p.? variant; symbols with gray filling—clinically affected individuals carrying the p.Gly234Arg variant only. c1 to c5—patients described in this report; XX y—current (2023) age in years; 1/2 below symbols—indicating the compound heterozygous state for both variants, 1/wt—indicating the heterozygous state for the p.Gly234Arg variant. Individuals without these symbols did not undergo molecular genetic analysis
Table 1 Summary of the clinical characteristics of patients c1 to c5Table 2 Characteristics of the variants in the calpain 3 gene identified in this studyPatient c1, now 65 years old, reported to our clinic in 1998 (aged 41 years) with slowly progressive weakness at that time mainly affecting her shoulder girdle without involvement of facial muscles. She had been a sportive child and played handball until the age of ~35 years. A muscle biopsy showed signs of a “very discrete unspecific myopathy.” Protein and enzyme studies of the muscle biopsy not including calpain 3 were negative as well as the molecular genetic analysis for facioscapulohumeral muscular dystrophy (FSHD, 4q form). The weakness was slowly progressive and increasingly also affected the legs. The current neurologic exam showed a predominantly proximal paresis affecting the arms more than the legs (Table 1). The patient is still able to walk a few steps but is wheelchair-bound for any longer distance. Creatine kinase (CK) concentration in serum was elevated to ~600 U/l. Electromyogram (EMG) examination showed myopathic changes in the quadriceps femoris, biceps brachii, and deltoid muscles. Motor and sensory nerve conduction studies were normal. Muscle ultrasound revealed hyperechogenicity in the deltoid and biceps brachii but not in distal arm muscles or muscles of the lower extremities. Diagnostic exome analysis revealed compound heterozygosity for a missense variant (NM_000070.3: c.700G>A, p.Gly234Arg) and an intronic variant (NM_000070.3: c.1746-20C>G, p.?) in CAPN3. No additional rare variants of likely clinical relevance were detected in genes previously associated with the patient’s clinical features.
Patient c2, now 64 years old, trained as a carpenter and bricklayer. In his 30s, he developed progressive problems working overhead caused by proximal weakness of the arms, initially diagnosed as brachial plexus neuritis. Later additional proximal leg weakness prompted the diagnosis of myopathy. A muscle biopsy showed—as in his sister—signs of an unspecified myopathy. The patient is still ambulant but cannot get up from a squatting position. The neurologic examination showed predominantly proximal pareses, somewhat milder than in his 1-year older sister c1, and the CK concentration was elevated at around 900 U/l. Muscle biopsy, EMG, or muscle MRI was not performed. Diagnostic Sanger sequencing of the two variants found in his sister c1 showed that he carries the same variants.
Patient c3, now 59 years of age, played handball in his youth. In his 20s, he developed extremely severe myalgia during and after training eventually leading to his abandonment of the sport. Exertional myalgia progressed over the years and became so severe that he had to retire from his job as a truck driver at the age of 41 years. Currently, patient c3 also complains of proximal arm weakness but the neurologic exam did not show any paresis. The CK concentration without prior exertion was normal (~70 U/l). EMG examination showed myopathic changes mainly in the biceps brachii and deltoid muscles but mild changes in the form of polyphasic potentials also in the quadriceps femoris and anterior tibial muscles. Diagnostic Sanger sequencing of the familial variants showed that he carries only the p.Gly234Arg missense variant.
Patient c4, now 56 years old, was also a sportive girl playing handball. However, she reports that her gait has always been “a little bit waddling” and her “shoulder blades protruded.” She noticed a weakness in her arms around the age of 40 years, subsequently also affecting her legs. She is now able to walk 50 m with a walking stick but requires a wheelchair for longer distances. Neurologic examination showed predominantly proximal pareses of approximately the same severity as her 9 years older sister c1. CK concentration was elevated to ~300–1000 U/l. Muscle biopsy, EMG, or muscle MRI was not performed. Diagnostic Sanger sequencing of the familial variants showed that she carries both variants.
Patient c5, now 45 years of age, is the son of patient c1 and suffers since age ~40 from severe generalized exertional myalgia without pareses elicited even by very mild exercise. He works as a sea freight inspector, now part-time due to his disease. The neurologic examination was normal and the CK concentration without prior exertion with 137 U/l in the normal range. EMG examination of the biceps brachii and quadriceps muscles was normal. Diagnostic Sanger sequencing of the familial variants revealed heterozygosity for the p.Gly234Arg missense variant.
None of the patients exhibited so far facial weakness, speech or swallowing problems, respiratory problems, or cardiac involvement. Distal pareses were mild or absent.
Comments (0)