
Remember me



The global prevalence of hypertension in pregnancy is increasing rapidly, severely affecting up to 10% of pregnant women worldwide [1]. Hypertension in pregnancy can cause severe complications in the second half of pregnancy, including prematurity, stillbirth, and growth restriction, and affects children's future growth and health [2,3]. Hypertension in pregnancy is likewise a major cause of morbidity, mortality, and hospitalization for both mothers and their infants [4]. Multiple pregnancies, parity, and history of preeclamptic pregnancies are all risk factors for hypertension in pregnancy, along with some cardiovascular risk factors, such as obesity, hypertension, type 2 diabetes, and hyperlipidemia [3]. Traditional risk factor such as obesity – BMI greater than 35 kg/m2 were also been identified [5,6].
Childhood obesity is a major public health problem, US childhood obesity rate is reported to be as high as 12.7% [7]. Childhood obesity can easily persist into adult obesity, thereby increasing cardiovascular morbidity and mortality, as well as the occurrence of diabetes, hypertension, cancer, and other adverse cardiometabolic alterations [8]. Obesity in adults is associated with hypertension in pregnancy events in both observational cohort studies [9,10] and Mendelian randomization studies [11]. However, few studies have investigated whether childhood obesity similarly affects hypertension in pregnancy. Therefore, it is unclear whether childhood obesity contributes to the development of hypertension in pregnancy.
Mendelian randomization analysis is a traditional statistical method for assessing the causal relationship between a risk factor and disease using the genetic variation of the risk factor as a tool [12]. This method dramatically reduces potential unmeasured confounders and reverse causality by independently isolating and randomly assigning at conception [13]. In this present study, we used Mendelian randomization approaches to assess whether childhood obesity is associated with hypertension in pregnancy.
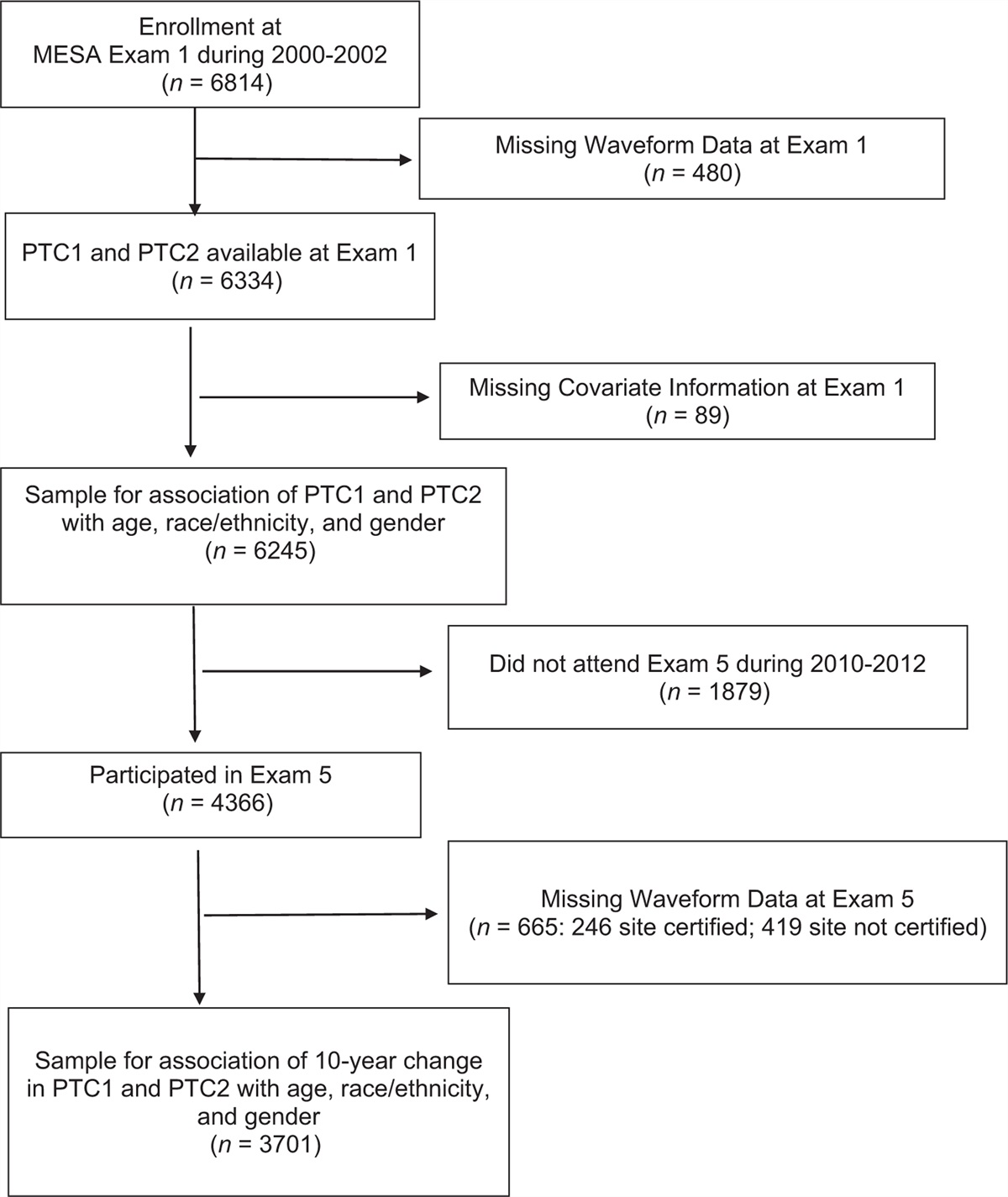
METHODS Study designThe Mendelian randomization design method uses publicly available datasets from extensive genome-wide association studies (GWAS) for risk factors and disease to examine whether exposure has a causal effect on disease emergence. Genetic variation is viewed as an instrumental variable in the Mendelian randomization design method. Mendelian randomization methods can overcome unmeasured confounding factors, allowing for more robust causal inferences. The Mendelian randomization design was based on three assumptions: genetic variation was strongly related to exposure; genetic variation was independent of other confounding factors; genetic variation was only related to outcome through the investigated exposure. We obtained publicly available summarized results from published studies, which have obtained institutional review board approval in their respective studies. No additional ethics approval was needed. We used two-sample Mendelian randomization [14,15] to research the causal relationship between childhood obesity and hypertension in pregnancy (Fig. 1).
 FIGURE 1:
FIGURE 1: Mendelian randomization model of childhood obesity and hypertension in pregnancy. Single-nucleotide polymorphisms (SNPs) was obtained, which associated with childhood obesity and the corresponding effect for these SNPs was estimated based on the risk of hypertension in pregnancy obtained from a large cohort of European population.
Data sourcesThe genetic instruments of childhood obesity, including 5530 cases and 8318 controls in European children, were obtained from the Early Growth Genetics (EGG) Consortium, which contained 2 442 739 single-nucleotide polymorphisms (SNPs) [16].
The GWAS-aggregated data for hypertension in pregnancy was obtained from the FinnGen project, a global research project in Europe, available at https://gwas.mrcieu.ac.uk/datasets/finn-a-HYPTENSPREG/.
We obtained five SNPs associated with childhood obesity from a preliminary meta-analysis, according to the threshold requirement of P less than 5 × 10−8 at the genome-wide significance level, As at least 10 instrumental variables are required for a Mendelian randomization study [17], we selected instrumental variables of value P less than 5 × 10−6[18,19] to obtain 14 SNPs for Mendelian randomization analysis. The parameters used to eliminate linkage disequilibrium among variables were kb = 10 000 and r2 = 0.01. The F statistic is used to estimate sample overlap effects and weak instrumental bias, and an F value greater than 10 is strong enough to limit bias from weak instrumental variables [20]. The SNP rs1040070 was removed because of the palindrome with medium-allele frequency. Details of the 14 finalized instrumental variables are listed in Table 1.
TABLE 1 - The detailed information of finalized single-nucleotide polymorphisms in exposure and outcomes. Exposure (childhood obesity ) Outcome (HDP) SNP EA OA SE Beta P value EAF SE Beta P value EAF F rs1040070 C G 0.027 −0.149 2.77E-08 NA 0.014 −0.024 0.084 NA 30.8 rs10913469 C T 0.033 0.177 7.99E-08 NA 0.018 0.527 0.004 NA 28.9 rs13130484 T C 0.027 0.143 1.30E-07 NA 0.014 0.007 0.608 NA 27.8 rs17697518 T C 0.039 0.186 1.85E-06 NA 0.019 0.018 0.362 NA 22.7 rs256335 T C 0.026 0.121 3.72E-06 NA 0.014 0.014 0.296 NA 21.5 rs28636 T C 0.032 −0.147 3.07E-06 NA 0.016 −0.039 0.015 NA 21.8 rs4833407 A C 0.027 0.123 3.88E-06 NA 0.014 0.011 0.409 NA 21.4 rs4854344 T G 0.035 0.245 3.22E-12 NA 0.019 0.028 0.134 NA 48.5 rs4864201 C T 0.028 −0.136 1.41E-06 NA 0.014 0.297 0.297 NA 23.3 rs571312 A C 0.031 0.199 1.25E-10 NA 0.018 0.009 0.574 NA 41.3 rs6752378 A C 0.026 0.170 1.05E-10 NA 0.014 0.027 0.053 NA 41.9 rs7138803 A G 0.027 0.167 6.50E-10 NA 0.014 0.024 0.089 NA 38.1 rs9299 T C 0.028 0.134 1.91E-06 NA 0.015 0.042 0.004 NA 22.7 rs9568856 A G 0.040 0.191 1.36E-06 NA 0.021 0.018 0.091 NA 23.4 rs9941349 T C 0.027 0.198 1.16E-13 NA 0.009 0.003 0.899 NA 54.9EA, effect allele; EAF, effect allele, frequency; HDP, hypertension in pregnancy; NA, not applicable; OA, other allele; SE, standard errors; SNP, single-nucleotide polymorphisms.
We used a two-sample Mendelian randomization analysis to estimate the direct effect of childhood obesity on the risk of hypertension in pregnancy. Mendelian randomization analysis was conducted using the inverse-variance-weighted (IVW) model, weighted-median estimator (WME), and Mendelian randomization-Egger regression. IVW was used as the primary method of Mendelian randomization analysis to assess the causal effects of childhood obesity with hypertension in pregnancy [21]. The leave-one-out sensitivity test consists of eliminating SNPs to determine the sensitivity of individual SNPs in this Mendelian randomization study [22]. The difference between various instrumental variables was analyzed by Cochran's Q (heterogeneity) test [23]. All of the data were analyzed using R (version 4.0.3) software (R Foundation for Statistical Computing, Vienna, Austria).
RESULTS Results of the Mendelian randomization study testing causal associationThree Mendelian randomization methods, including IVW, Mendelian randomization-Egger, and weighted median regression, were used to investigate the causal effects of obesity on the risk of hypertension in pregnancy (Figs. 2 and 3). IVW method suggests that childhood obesity positively correlates with hypertension in pregnancy [IVW Mendelian randomization odds ratio (OR) = 1.161, 95% CI 1.086–1.239, P = 9.92 × 10–6]. Similar results were observed using the weighted median method [weighted median OR = 1.123, 95% CI 1.038–1.215, P = 0.04]. No significant causal relationship in MR-Egger analysis (OR = 1.279, 95% CI 0.906–1.805; P = 0.187). The causal effects of each genetic variant on hypertension in pregnancy are shown in Figs. 2 and 4.
 FIGURE 2:
FIGURE 2: Scatter plot to visualize causal effect of childhood obesity on the risk of hypertension in pregnancy. The slope of each line corresponds to the causal estimate using different MR methods. IVW, inverse-variance weighted; MR, Mendelian randomization.
 FIGURE 3:
FIGURE 3: Forest plot to visualize causal effect of each single single-nucleotide polymorphisms on the risk of hypertension in pregnancy.
 FIGURE 4:
FIGURE 4: Forest plot to visualize the effect estimates presented as odds ratio of hypertension in pregnancy per 1-unit-higher log-odds of childhood obesity.
Analysis of horizontal pleiotropyFunnel plots display the individual Wald ratios for each SNP plotted against their precision, where asymmetry indicates directional horizontal pleiotropy (Fig. 5). The horizontal pleiotropy of a small number of instrumental variables is difficult to assess using funnel plots. For this reason, in order to investigate the direction of the horizontal pleiotropy, Mendelian randomization-Egger intercepts were utilized; however, no directional pleiotropy was found (P = 0.58).
 FIGURE 5:
FIGURE 5: Funnel plots to visualize overall heterogeneity of Mendelian randomization estimates for the effect of childhood obesity on the risk of hypertension in pregnancy.
The effects of individual genetic instruments on pregnancy hypertensionWe also performed a leave-one-out analysis and found that the causal association still existed even when a single SNP was eliminated one by one (Fig. 6). Consequently, the estimated effects cannot be explicated by any single genetic instrument.
 FIGURE 6:
FIGURE 6: Leave-one-out plot to visualize causal effect of childhood obesity on the risk of hypertension in pregnancy when leaving one single-nucleotide polymorphism out.
DISCUSSIONOn the basis of Mendelian randomization framework, our study showed that childhood obesity contributes significantly to the risk of hypertension in pregnancy. A sensitivity analysis confirmed the robustness of the results, suggesting a relationship between childhood obesity and pregnancy hypertension independent of confounding factors. The odds ratio of 1.161 (95% CI 1.086–1.239) indicates a substantial causal relationship between childhood obesity and hypertension during pregnancy.
Childhood obesity was found to be a risk factor for hypertension, cardiovascular and cerebrovascular diseases, diabetes, and a host of other health problems, according to previous observational studies [24]. The incidence of pregnancy hypertension with childhood overweight and childhood abdominal obesity was examined in a nearly 20-year cohort study with 3412 female participants; results revealed a higher risk of childhood adiposity with an increased risk of pregnancy hypertension [childhood overweight, relative risk (RR) = 1.66, 95% CI 1.07–2.52; abdominal obesity RR = 2.55, 95% CI 1.34–4.85] [5]. Another cohort study of 49 600 nulliparous women indicated that BMI at ages 7 and 13 years increased above the average level, and so did the risks of gestational hypertension and preeclampsia, as BMI increased above average, RR for hypertension in pregnancy was 1.66 (95% CI 1.42–1.94) and for preeclampsia was 1.57 (95% CI 1.46–1.70) [6]. These findings revealed that childhood obesity might be an independent risk factor for pregnancy-related hypertension. According to Mendelian randomization research, children who are obese have an increased risk of developing essential hypertension [5]; furthermore, another Mendelian randomization study discovered a direct link between pregnancy-related hypertension issues and obesity [11].
Our Mendelian randomization study provides the first comprehensive assessment of the causal relationship between childhood obesity and hypertension in pregnancy. In contrast to conventional observational epidemiological studies, Mendelian randomization analysis can offer the most vital support for the causal relationship between childhood obesity and hypertension in pregnancy, potentially overcoming unmeasured confounders and reverse causation that could skew an observational study. The findings of observational studies were supported by the causation estimates for childhood obesity and hypertension in pregnancy obtained using Mendelian randomization analysis. To identify the causal relationship, we identified 14 SNPs using two GWAS datasets and three different models, including IVW, weighted median, and Mendelian randomization-Egger regression. Obesity was identified as a risk factor for hypertension in pregnancy by IVW analysis, with a 1 SD increase in obesity resulting in a 16% increased risk of hypertension in pregnancy, as determined by Mendelian randomization-Egger and weighted median regression. The importance of preventing childhood obesity in preventing hypertension in pregnancy is highlighted by our findings, which also provide genetic information that can be used to understand better the pathogenesis of the emergence of hypertension in pregnancy.
The biochemical mechanisms through which childhood obesity influences the risk of hypertension in pregnancy are still unknown. Insulin resistance [25], inflammation upregulation [26], oxidative stress [27], and endothelial dysfunction [28] are all possible explanations for the metabolic alterations associated with childhood obesity. Much evidence suggests that hypertension in pregnancy may be associated with insulin resistance. As in patients with essential hypertension, hypothesized mechanisms by which insulin resistance may increase blood pressure in pregnancy include sympathetic nervous system activation, renal sodium retention, increased cation transport, and associated endothelial dysfunction [29]. In addition, hyperinsulinemia and insulin resistance, which are commonly seen in obese individuals, also contribute to hypertension development. Hyperinsulinemia may increase sympathetic nervous system activity, activate the renin–angiotensin system, and cause renal sodium retention, which may increase blood pressure if sustained [30]. Thus the mechanism of childhood obesity-related postadult hypertension in pregnancy may be related to insulin resistance, and further mechanisms deserve to be confirmed by more studies.
One of our study's strengths is that we use a two-sample Mendelian randomization design to evaluate the causal relationships between childhood obesity and hypertension in pregnancy, which reduces the potential of bias from confounding. A further strength is that our study of hypertension in pregnancy was restricted to people with European ancestry is another advantage because it may have lessened population stratification bias.
Nonetheless, our study has certain limitations. First, the participants included in our study all came from the European ancestry GWAS database. Therefore, the reliability of the causal associations should be validated in other populations. Second, only four SNPs that matched the P less than 5 × 10–8 bioinformatics cutoff were chosen. The number of SNPs would make matching instrumental variables in the results difficult, but it would also weaken any association. Due to this reason, we chose SNPs with a less stringent significance threshold of 5 × 10–6, as suggested in earlier studies [19,31], with the disadvantage of causing weak instrumental variable bias. To assess the risk of such bias, we calculated F statistics, and all of them had an F value greater than 10. Third, the publicly available GWAS of childhood obesity did not include information on the characteristics of childhood obesity, such as weight, height, and abdominal circumference. These factors may be helpful in further classifying childhood obesity to improve accuracy while controlling for potential confounding variables. Fourth, hypertension in pregnancy is divided into four categories: chronic hypertension, gestational hypertension, preeclampsia, and chronic hypertension superimposed on preeclampsia. In our article, we found that childhood obesity is associated with postadult hypertension in pregnancy, but no subgroup data were available to identify, which type of hypertension in pregnancy is linked to childhood obesity. Subgroup analyses will be possible when the GWAS data on the classification of hypertension in pregnancy are available in the future in order to clarify the relationship between childhood obesity and different types of hypertension in pregnancy.
In conclusion, our two-sample Mendelian randomization analysis provides genetic support for the hypothesis that childhood obesity is causal in the etiology of hypertension in pregnancy. Childhood obesity was linked to an increased risk of hypertension in pregnancy. The findings demonstrate the need for specialized clinical care for people with a history of childhood obesity to prevent hypertension in pregnancy.
ACKNOWLEDGEMENTSWe want to acknowledge the participants and investigators of Early Growth Genetics (EGG) Consortium and FinnGen study for sharing the genetic data.
Author contributions: B.H., as the first author performed data analysis and wrote the manuscript, J.S. and Y.S. conceived of the study idea. X.H. developed the theory and performed the computations. F.L. contributed suggestions for verifying the analytical methods and revised the manuscript. F.L. provided advice on experimental design and finalized the manuscript. All authors discussed the results and contributed to the final manuscript.
Conflicts of interestThere are no conflicts of interest.
REFERENCES 1. Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J, Blomstrom-Lundqvist C, Cifkova R, De Bonis M, et al. ESC Scientific Document Group. 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy. Eur Heart J 2018; 39:3165–3241. 2. Odegard RA, Vatten LJ, Nilsen ST, Salvesen KA, Austgulen R. Preeclampsia and fetal growth. Obstet Gynecol 2000; 96:950–955. 3. Agrawal A, Wenger NK. Hypertension during pregnancy. Curr Hypertens Rep 2020; 22:64. 4. Lu CQ, Lin J, Yuan L, Zhou JG, Liang K, Zhong QH, et al. Pregnancy induced hypertension and outcomes in early and moderate preterm infants. Pregnancy Hypertens 2018; 14:68–71. 5. He Y, Tian J, Blizzard L, Oddy WH, Dwyer T, Venn AJ. Associations of childhood adiposity and changes in adiposity status from childhood to adulthood with pregnancy hypertension. Pregnancy Hypertens 2020; 19:218–225. 6. Pedersen DC, Bjerregaard LG, Nohr EA, Rasmussen KM, Baker JL. Associations of childhood BMI and change in BMI from childhood to adulthood with risks of hypertension in pregnancy. Am J Clin Nutr 2020; 112:1180–1187. 7. Martin FZ, Fraser A, Zuccolo L. Alcohol intake and hypertensive disorders of pregnancy: a negative control analysis in the ALSPAC Cohort. J Am Heart Assoc 2022; 11:e025102. 8. Horesh A, Tsur AM, Bardugo A, Twig G. Adolescent and childhood obesity and excess morbidity and mortality in young adulthood-a systematic review. Curr Obes Rep 2021; 10:301–310. 9. Bicocca MJ, Mendez-Figueroa H, Chauhan SP, Sibai BM. Maternal obesity and the risk of early-onset and late-onset hypertensive disorders of pregnancy. Obstet Gynecol 2020; 136:118–127. 10. Rantakallio JSS, Nevalainen JE, West SI, Ollila MM, Puukka K, Bloigu AH, et al. Association of self-reported polycystic ovary syndrome, obesity, and weight gain from adolescence to adulthood with hypertensive disorders of pregnancy: a community-based approach. Hypertension 2021; 77:1010–1019. 11. Wang W, Tan JS, Hua L, Zhu S, Lin H, Wu Y, et al. Genetically predicted obesity causally increased the risk of hypertension disorders in pregnancy. Front Cardiovasc Med 2022; 9:888982. 12. Smith GD, Ebrahim S. ’Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 2003; 32:1–22. 13. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 2014; 23:R89–R98. 14. Lawlor DA. Commentary: two-sample Mendelian randomization: opportunities and challenges. Int J Epidemiol 2016; 45:908–915. 15. Richmond RC, Hemani G, Tilling K, Davey Smith G, Relton CL. Challenges and novel approaches for investigating molecular mediation. Hum Mol Genet 2016; 25:R149–R156. 16. Bradfield JP, Taal HR, Timpson NJ, Scherag A, Lecoeur C, Warrington NM, et al. Early Growth Genetics Consortium. A genome-wide association meta-analysis identifies new childhood obesity loci. Nat Genet 2012; 44:526–531. 17. Savage JE, Jansen PR, Stringer S, Watanabe K, Bryois J, de Leeuw CA, et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet 2018; 50:912–919. 18. Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 × 2 factorial Mendelian randomization study. J Am Coll Cardiol 2015; 65:1552–1561. 19. Zou XL, Wang S, Wang LY, Xiao LX, Yao TX, Zeng Y, et al. Childhood obesity and risk of stroke: a Mendelian randomisation analysis. Front Genet 2021; 12:727475. 20. Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol 2011; 40:740–752. 21. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015; 44:512–525. 22. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol 2017; 32:377–389. 23. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016; 40:304–314. 24. Lawlor DA, Leon DA. Association of body mass index and obesity measured in early childhood with risk of coronary heart disease and stroke in middle age: findings from the Aberdeen children of the 1950s prospective cohort study. Circulation 2005; 111:1891–1896. 25. Thota P, Perez-Lopez FR, Benites-Zapata VA, Pasupuleti V, Hernandez AV. Obesity-related insulin resistance in adolescents: a systematic review and meta-analysis of observational studies. Gynecol Endocrinol 2017; 33:179–184. 26. Rainone V, Schneider L, Saulle I, Ricci C, Biasin M, Al-Daghri NM, et al. Upregulation of inflammasome activity and increased gut permeability are associated with obesity in children and adolescents. Int J Obes (Lond) 2016; 40:1026–1033. 27. Kilic E, Özer ÖF, Erek Toprak A, Erman H, Torun E, Kesgin Ayhan S, et al. Oxidative stress status in childhood obesity: a potential risk predictor. Med Sci Monit 2016; 22:3673–3679. 28. Bruyndonckx L, Hoymans VY, Lemmens K, Ramet J, Vrints CJ. Childhood obesity-related endothelial dysfunction: an update on pathophysiological mechanisms and diagnostic advancements. Pediatr Res 2016; 79:831–837. 29. da Silva AA, do Carmo JM, Li X, Wang Z, Mouton AJ, Hall JE. Role of hyperinsulinemia and insulin resistance in hypertension: metabolic syndrome revisited. Can J Cardiol 2020; 36:671–682. 30. El Meouchy P, Wahoud M, Allam S, Chedid R, Karam W, Karam S. Hypertension related to obesity: pathogenesis, characteristics and factors for control. Int J Mol Sci 2022; 23:12305. 31. Cao Z, Wu Y, Li Q, Li Y, Wu J. A causal relationship between childhood obesity and risk of osteoarthritis: results from a two-sample Mendelian randomization
Comments (0)