
Remember me
Coronaviruses are single-stranded RNA viruses, which belong to the family Coronaviridae. The family consists of four genera: α, β, γ, and δ. There are seven pathogenic to humans coronaviruses: SARS-CoV-2, SARS-CoV, MERS-CoV, HCoV-HKU1, HCoV-OC43, HCoV-NL63, and HCoV-229E. HCoV-NL63 and HCoV-229E belong to α-coronaviruses, the rest are considered β-coronaviruses. HCoV-HKU1, HCoV-OC43, HCoV-NL63, and HCoV-229E usually cause mild or moderate respiratory diseases, while SARS-CoV-2, SARS-CoV, and MERS-CoV cause severe respiratory diseases. The viruses differ in their replication potential, with SARS-CoV-2 virus outperforming other viruses, and having high spreading potential. SARS-CoV caused severe acute respiratory syndrome (SARS) outbreak in 2003 leading to over 8000 cases with fatality rate of 11% (World Health Organization, 2003). The Middle East Respiratory Syndrome (MERS) is a viral respiratory disease caused by the Middle East Respiratory Syndrome Coronavirus (MERS‐CoV). This virus was identified in Saudi Arabia in 2012 (Bermingham et al., 2012). MERS-CoV can cause a variety of symptoms and сlinical manifestations that range from mild to severe, including acute respiratory distress syndrome (ARDS) and organ failure. The latter two conditions are often associated with uncontrolled cytokine production, leading to a cytokine storm. According to WHO, the mortality rate among cases of MERS-CoV disease was 36% (WHO, 2024), which is higher compared to both SARS-CoV and SARS-CoV-2. In comparison, the mortality rate of SARS-CoV-2 and SARS-CoV varies between 2% and 10% (Meo et al., 2020). In contrast, HCoV-NL63, OC43, 229E, HKU1 infections are associated with mild respiratory tract diseases, and there is no extensive data on mortality among those infected. A retrospective cohort study, conducted from October 2012 to December 2017, investigated adults infected with HCoV-229E and HCoV-OC43 coronaviruses, and suggested that HCoV-229E is more virulent compared to HCoV-OC43. The study reported a 30-day all-cause mortality rate of 25% for patients infected with HCoV-229E and 9.1% for HCoV-OC43- infected adult patients (Choi et al., 2021).
There are multiple factors that contribute to the MERS pathogenicity which potentially could explain its higher mortality rate. Among those factors are difference in structural proteins as well as in accessory proteins. For example, MERS is the only coronavirus that utilizes dipeptidyl peptidase IV (DPP4) or CD26 for cellular entry (Wang et al., 2013). Such difference in the target receptor is due to difference in the receptor binding domain of structural protein S (Wang et al., 2013). Within S protein of MERS-CoV there are two furin-cleavage sites which facilitate virus entry into the host cell. Besides this, multiple studies demonstrate that MERS accessory protein ORF4a is a potent inhibitor of antiviral stress response (Menachery et al., 2017; Rabouw et al., 2016). Together with ORF4a, accessory proteins ORF4b, ORF5, and structural proteins M and N are potent inhibitors of interferon (Yang et al., 2013; Chang et al., 2020).
The severe disease manifestation seen following infection with MERS, SARS-CoV, and SARS-CoV-2 could be explained by massive cytokine storm triggered by viruses. All three viruses infect airway epithelial cells, where they replicate. MERS-CoV, besides airway epithelial cells, was also detected within human T cells where it was shown to cause apoptosis (Chu et al., 2016). Such ability to infect and cause apoptosis of T cells is not observed in other human coronaviruses, and can contribute to the difference seen in MERS-CoV pathogenesis. As reported by Liu et al. SARS-CoV-2, SARS-CoV and MERS-CoV could infect dendritic cells, mononuclear macrophages and other peripheral blood mononuclear cells, inducing the cells to release large amounts of cytokines and chemokines (Liu et al., 2021; Zhou et al., 2015). Together with infection of dendritic cells, mononuclear macrophages and other peripheral blood mononuclear cells, MERS-CoV virus was suggested to be replicated in them, further causing apoptosis and abnormal cytokine release from infected cells (Lau et al., 2013). This sets MERS-CoV virus apart from other coronaviruses. In summary, it is possible that MERS-CoV’s higher mortality are due to unique properties of the virus, such as replication in immune cells, leading to their apoptosis, delayed interferon response, and abnormal cytokine release. High levels of IL-6, IP-10, IL-8, RANTES, and IFN-α were detected in the serum of patients with severe MERS compared to those with mild MERS (Min et al., 2016; Kim et al., 2016). Serological studies of patients with MERS-CoV have shown that low levels of IFN-alpha secretion were observed in patients with severe disease up to death, while low levels of type I IFN correlate with recovery and positive outcome (Faure et al., 2014). Factors such as the time interval between the onset of symptoms and hospitalization, age, and comorbidities influence the further course of the disease, up to and including a fatal cytokine storm (Ahmadzadeh et al., 2020).
Overall, the differences in the viral infection outcome are due to difference in viruses’ structure and function. This review summarizes similarities and highlights differences in the structure and function of structural proteins in all seven human coronaviruses.
The genome of all seven coronaviruses is similar in organization. It contains coding regions for four structural proteins (spike glycoprotein (S), nucleocapsid phosphoprotein (N), membrane protein (M), and envelope (E) protein), 15 non-structural proteins (nsp) and 7 accessory proteins (Brant et al., 2021). The detailed summary of structural features of four structural proteins is provided along with their main functions in viral lifecycle as well as in host cell physiology.
2 Structure and function of S protein2.1 StructureFor all seven types of human coronaviruses, the spike protein is a key structural element that plays a crucial role in viral infection. S proteins form large crown–like spikes on the surface of the virus, the feature that gave the name of the taxonomic group of viruses - coronaviruses. In coronaviruses, including SARS-CoV-2, SARS-CoV, MERS-CoV, HCoV-HKU1, HCoV-OC43, HCoV-NL63 and HCoV-229E, the spike protein acts as a type I transmembrane fusion glycoprotein. During viral entry, the spike protein interacts with the protein receptors of the host cell and is split into two functional subunits: S1 and S2. Depending on the coronavirus type, the S1/S2 spike protein site is cleaved either by furin in the infected cell or by host cell proteases during viral entry (Kirchdoerfer et al., 2016; Hoffmann et al., 2020; Shang et al., 2020). The presence of multiple arginine residues at the S1/S2 site is believed to make the protein more susceptible to cleavage. Besides SARS-CoV-2, three (MERS-CoV, HCoV-OC43 and HCoV-HKU1) of the six other HCoVs have furin cleavage sites (Liu et al., 2021). Within SARS-CoV-2 virus the furin-cleavage site is distinguished by four amino acids: 681-PRRA-684. Knockout of furin with CRISPR-Cas9 showed a significant reduction in cleavage in the S1/S2 region of SARS-CoV-2 spike glycoprotein, but did not completely prevent this process (Wrapp et al., 2020; Papa et al., 2021; Ou et al., 2020). In vitro studies of the S1/S2 region of the SARS-CoV-2 spike protein highlighted the importance of residue R683 in the RRAR motif for furin recognition. Besides this, serines at the edges of this motif, specifically S680 and S686, can be phosphorylated by basophilic and proline-directed kinases, which negatively affects furin cleavage at this site (Örd et al., 2020). Substituting proline with arginine at residue 681 disrupts the proline motif necessary for phosphorylation, which in turn increases the potential for furin cleavage at this site (Beaudoin et al., 2022).
It is worth mentioning here that MERS-CoV virus is a unique in its structure compared to other human coronaviruses as far as it has two furin-recognition motifs (RXXR): at S1/S2 border and at S2’ site. This factor was suggested to contribute to high pathogenicity of MERS-CoV (Millet and Whittaker, 2014; Wu and Zhao, 2020).
As far as S protein has to be proteolytically activated in order to be able to fuse with host cell membrane, it is of interest to focus on proteases that activate coronaviruses without furin-recognition site. The SARS-CoV S protein contains several sites that are subject to cleavage, but the major furin cleavage site has not been identified. For SARS-CoV it was shown that endosomal cysteine protease – cathepsin L plays role in membrane fusion (Simmons et al., 2005). Besides cathepsin L, other proteases, such as elastase and coagulation factor Xa were shown to be active on SARS-CoV (Matsuyama et al., 2005; Du et al., 2007). The furin cleavage site is absent in HCoV-229E, while for HCoV-NL63 it is located in S2’ site (Wu and Zhao, 2020). Human coronavirus 229E uses various proteases, such as cathepsin L, TMPRSS2, and trypsin, to activate cell entry (Kawase et al., 2009). The I577S mutation in the spike protein allowed increased cathepsin utilization by the 229E virus, however, reduced cell replication ability was observed. This may indicate that the endosomal route of coronavirus entry is less preferable for 229E compared to activation via TMPRSS2 (Shirato et al., 2017; Bonnin et al., 2018).
Interestingly, peptides with amino-acid sequences which are similar to the furin cleavage-sites of original SARS-CoV-2 virus as well as its variants (Alpha, Delta, and Omicron) are potent inhibitors of α7 and α9α10 nicotinic acetylcholine receptors (Hone et al., 2024). Considering that both α7 and α9α10 receptors are abundantly expressed in lung epithelial cells (Hollenhorst and Krasteva-Christ, 2021), it could be hypothesized that the receptors are used by SARS-CoV-2 as an alternative to ACE2 entry point to the host cell. α7 and α9α10 receptors are also expressed by immune cells, where they modulate cytokine release (Fujii et al., 2017). It is possible that these receptors contribute to the immune response observed in SARS-CoV-2 infection.
The S1 subunit is responsible for primary contact, while the highly conserved S2 subunit promotes membrane fusion between the host cell and the virus (Heald-Sargent and Gallagher, 2012; Holmes et al, 1981; Perlman and Netland, 2009; Künkel and Herrler, 1993; Qian et al., 2013). The S1 subunit contains the N-terminal domain (NTD), and C-terminal domain (CTD), harboring receptor-binding domain (RBD) and receptor-binding motif (RBM) (Peng et al., 2021) in all but HCoV-OC43 virus (Figure 1 and Supplementary Table 1). According to Caetano-Anolles et al. the NTD is characterized by a galectin-like structure, a common target of mutations, which helps the virus evade the physiological responses of the host (Guruprasad, 2021; Caetano-Anollés et al., 2022).
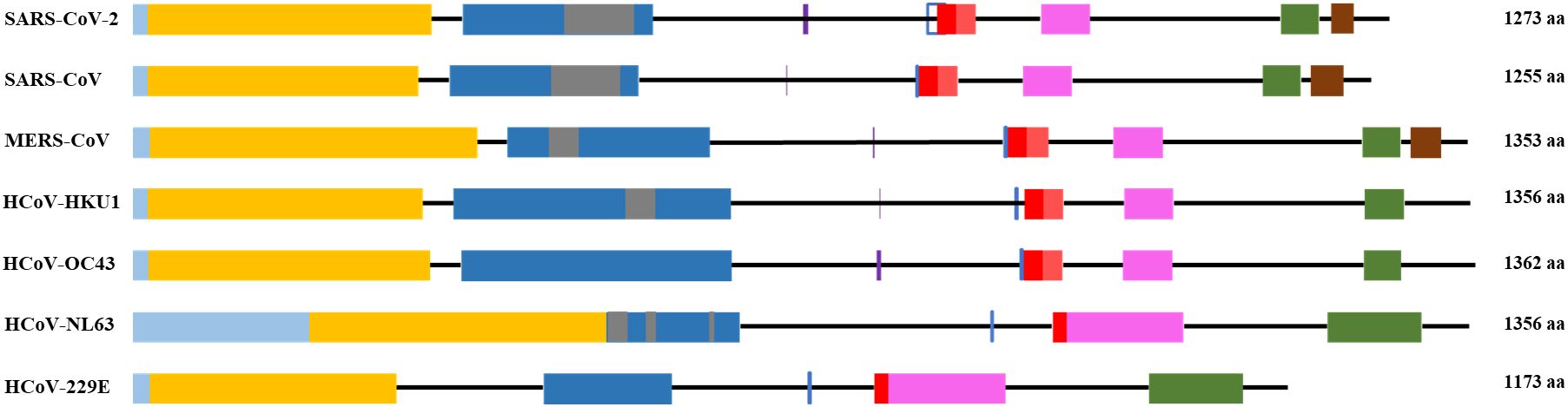
Figure 1. General scheme of the S protein structure. Following color code is used: light blue- signal sequence, orange - N-terminus domain of S1 subunit, blue – C-terminus domain of S1 subunit, grey – receptor binding motif, red – fusion peptide, pink – heptad repeat 1, green – heptad repeat 2, brown – transmembrane domain. Purple vertical line - S1/S2 cleavage site, blue vertical line - S2’ cleavage site.
Mutations within S protein change the affinities of the RBD to the host receptor, and lead to emergence of new viral variants with increased infectivity and transmissibility. There are five variants of concern of SARS-CoV-2 that emerged during the COVID-19 pandemic: Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2) and Omicron (B.1.1.529). The alpha variant of SARS-CoV-2 has 23 mutations, 9 of which are within S protein. Mutations that change the biological processes of the virus include N501Y (increases binding to the target receptor), P681H (improves transmissibility) and D614G (Liu et al., 2022; Mohammadi et al., 2021; Yang et al., 2021). D614G mutation in the wild type of SARS-CoV-2 increased the infectivity and transmissibility of the virus (Daniloski et al., 2021). Also, the S1/S2 junction cleavability has been increased by the D614G mutation (Gobeil et al., 2021). The beta variant has three mutations in the RBD, these include K417N, E484K and N501Y, and six mutations in the remaining regions of the spike. Similarly to the N501Y alpha variant, the K417N and E484 substitutions improve the affinity of the virus for its target receptor, angiotensin-converting enzyme 2 (ACE2) (Funk et al., 2021). The gamma variant of SARS-CoV-2 has 17 nonsynonymous mutations, ten of which are located in the S protein genes. Among them K417T, E484K and N501Y mutations which help the virus to escape immune surveillance (Chen et al., 2021; Faria et al., 2021; Naveca et al., 2021; Wang et al., 2021). In the delta variant of SARS-CoV-2, 17 mutations were found. In S1, two deletions occurred: E156del and F157del (Chen et al., 2020; Tatsi et al., 2021). The P681R mutation increases the cleavability of the S1/S2 site, which leads to increased replication in the delta variant of SARS-CoV-2 (Takeda, 2022; Saito et al., 2022). The F306L mutation of the SARS-CoV-2 delta variant may lead to strengthened binding between ACE2 and spike protein, and this mutation has been reported to be associated with increased mortality during infection (Sila et al., 2024). In the omicron variant, 39 mutations were identified in the spike protein, 15 of which were found in the RBD region. The mutations Q498R and Q493K were reported to weaken the RBD-ACE2 interaction, while P681H resulted in high transmissibility of the virus (Kim et al., 2021).
Similarly to SARS-CoV-2, S protein of other human coronaviruses was shown to have mutations that influence the pathogenicity of the viruses. For example, using next generation sequencing, a subgenotype of the HCoV NL63 virus with the I507L mutation was identified, which enhanced the penetration of the virus into the host cells, and was associated with severe course of the lower respiratory tract disease (Wang et al., 2020). Point mutations H183R and Y241H in the spike protein of the HCoV-OC43 virus resulted in weakened protein synthesis and increased neuroinvasiveness of the virus, which caused apoptosis in mouse neuronal cells (Favreau et al., 2009).
The amino acid sequence of the S protein and RBD of SARS-CoV compared to SARS-CoV-2 is 76% and 74% identical, respectively (Jaimes et al., 2020). The RBD of the SARS-CoV-2 S protein is located between 319 and 541 residues. The SARS-CoV RBD is located between residues 306 and 526 with residues. It forms an extended tyrosine-rich loop and binds directly to the angiotensin-converting enzyme 2 (ACE2) receptor (Li et al., 2005). According to Li and colleagues, the region spanning 577-597 residues of the SARS-CoV RBD matches the S1 region of HCoV-NL63 (Li et al., 2007).
The RBD has a core subdomain characterized by a five-stranded antiparallel β-sheet (found in β-HCoV) or a six-stranded β-sandwich (found in α-HCoV) and a receptor-binding subdomain. The differences in the receptor-binding subdomain explain the affinities of the virus to the target receptor. For example, MERS-CoV has unique features within its receptor-binding subdomain, enabling it to bind to DPP4. Particularly, unlike SARS-CoV and SARS-CoV-2 which have extended loop between two short antiparallel β-strands, MERS-CoV has a β-sheet structure made up by four antiparallel β-strands (Wang et al., 2013).
In almost all HCoVs, the RBD was found in the CTD of the S1 subunit. Only OC43 is distinguished by the location of the RBD in the NTD. It is also known that the S1-NTD of the HCoV-HKU1 virus mediates primary attachment via glycan binding (Qian et al., 2015; Kirchdoerfer et al., 2016).
Following interaction of the RBD with the cell receptor, viral entry into the host cell requires the use of proteases to activate the spike protein (Bestle et al., 2020; Hoffmann et al., 2020).
The S2 subunit of the spike protein is highly conserved compared to the S1 subunit. S2 consists of fusion peptide (FP), heptad repeat 1 (HR1), heptad repeat 2 (HR2) and transmembrane domain (Figure 1). Interaction of the virus with the host cell leads to refolding of HR1 and releasing of the fusion peptide. FP mediates the fusion of the host cell membrane lipid bilayer with the viral membrane and further cell hijacking (Zhang et al., 2018; Heald-Sargent and Gallagher, 2012; Belouzard et al., 2012; Li, 2016; Huang et al., 2020; Xia et al., 2020). The comparative structures of human coronaviruses are presented in the Table 1.

Table 1. Structural comparison of S protein from different coronaviruses, top and side views.
According to the data provided by Song F. et al., using the pseudotyping method with Modified Vaccinia virus Ankara, it was determined that the full-length S protein of MERS-CoV, in its N-glycosylated state, is estimated to be 210 kDa. Subsequent investigations propose the cleavage of the mature full-length S glycoprotein into an amino-terminal domain (S1) and an approximately 85-kDa carboxy-terminal domain (S2), which is membrane-anchored. The cleavage occurs between 751 and 752 residues (Song et al., 2013).
SARS-CoV-2, SARS-CoV and MERS-CoV exhibit structural mimicry with several alternative receptors aside from primary ones. For example, all three have similar receptor binding motifs (RBMs) to complement Factor H and EGF-like domains. SARS-CoV showed mimicry for clitocypin-5 cysteine protease, von Willebrand factor, and intracellular adhesion molecule 5. The RBM of SARS-CoV-2 mimics TNF receptors, neuroserpin, IL-6 receptors, and ephrin-B2. MERS-CoV mimics TNF ligands, fibronectin type III, transferrin receptor protein 1, and toxoplasma gondii surface antigen 3. These structural resemblances imply possibility for alternative pathways through which coronaviruses could modulate host cell invasion, cellular metabolism, immune responses, and disease severity (Beaudoin et al., 2021).
The HCoV-HKU1 and HCoV-OC43 spike glycoproteins consist of 1356 and 1362 amino acids. Similarly to HCoV-OC43, HCoV-HKU1 utilizes 9-O-Acetylated-sialic acid as a receptor to engage the host cells and initiate infection (Millet and Whittaker, 2014; Huang et al., 2015; Li et al., 2020).
HCoV-NL63 and HCoV-229E belong to the alpha subgroup of coronaviruses. The HCoV-NL63 spike protein belongs to type I single-chain transmembrane glycoprotein, the molecular weight of which is estimated at 128-160 kDa before and 150-200 kDa after glycosylation. Comparison of the amino acid sequence of HCoV-NL63 with HCoV-229E, SARS-CoV-2 and SARS-CoV showed 50%, 17.1%, and 25% sequence identity, respectively. HCoV-NL63 shares ACE2 receptor binding with SARS-CoV-2 and SARS-CoV, and uses ACE2 as a target receptor required for cellular entry (Hofmann et al., 2005). A distinctive feature of HCoV-NL63 is that the N-terminus region of the spike protein contains 179 amino acids that have no homology with any of the HCoVs (Smith et al., 2006; Castillo et al., 2023; Pöhlmann et al., 2006; Brielle et al., 2020).
HCoV-229E is known to cause mild respiratory infections in humans (Hamre and Procknow, 1966). The specific receptor for HCoV-229E is aminopeptidase N (APN), also known as CD13. The spike protein binds to the APN receptor, initiating the attachment and fusion of the virus with the host cell membrane (Yeager et al., 1992). The findings by Blau et al. suggest that HCoV-229E undergoes endocytosis following the binding of the spike protein at the plasma membrane. Subsequently, the virion is sorted into endosomes, where fusion between the viral envelope and endocytic membrane takes place (Blau and Holmes, 2001).
2.2 Function2.2.1 Cellular entryThe most crucial function of spike proteins is the facilitation of cellular entry into the host cell via receptor binding and fusion (Pillay, 2020). The trimeric protein in SARS-CoV-2 viruses accomplishes that by binding to angiotensin-converting enzyme 2 of the host cell through the receptor binding domain within the S1 subunit. S protein of SARS-CoV-2 and MERS-CoV is cleaved into S1 and S2 proteins during viral biosynthesis in a host cell, then S2 protein is cleaved at S2’ site. While S1 binds the receptor, the S2 subunit acts as an anchor of the S protein to the membrane of the virus and facilitates membrane fusion.
SARS-CoV and SARS-CoV-2 can enter cells through non-endosomal and endosomal routes (Hofmann and Pöhlmann, 2004; Peng et al., 2021; Cesar-Silva et al., 2022; Simmons et al., 2005) (Figure 2). Non-endosomal route involves fusion of viral envelope with host cell membrane. The process of initiation of virion entry to the host cell occurs by binding of the spike protein to a receptor on the cell surface. The endosomal route relies on clathrin-mediated endocytosis. When compared to non-endosomal entry, endocytosis leaves no traces of viral proteins on the membrane, which allows the virus to evade detection by immune cells.
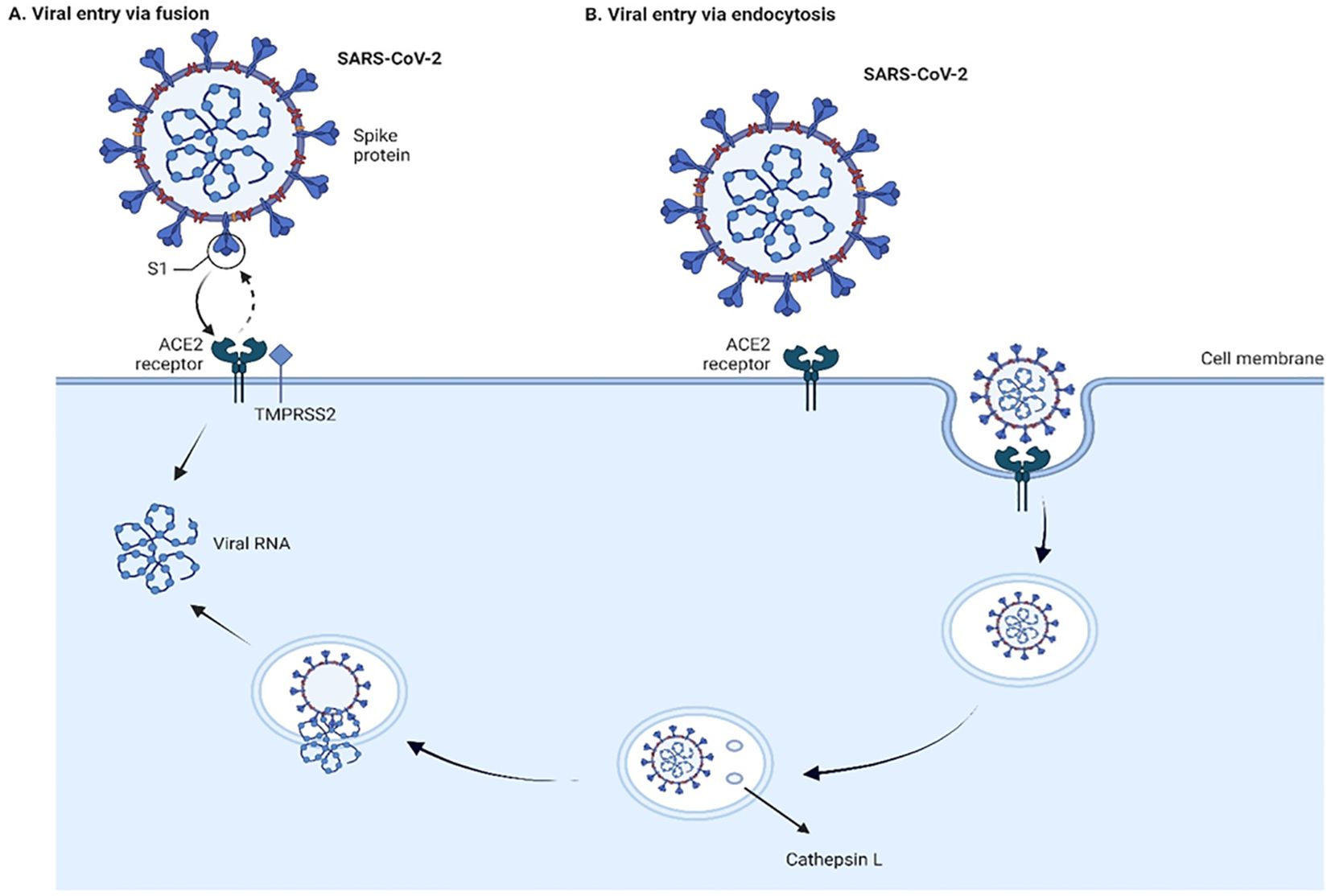
Figure 2. Viral entry into the cells through fusion (A) and endocytosis (B) Two pathways of cell entry are shown: endosomal and through fusion. The endosomal route is mediated in the case of a deficiency of transmembrane protease, serine 2 (TMPRSS2), and the virus-ACE2 complex internalizes. It does so through clathrin-mediated endocytosis, and the cleavage takes place inside the endosome via cathepsin L which requires a low pH environment. In the case of sufficient amount of TMPRSS2, the cleavage is done on the surface of the host cell membrane by TMPRSS2. The figure is created using BioRender.
Proteolytic cleavages of S protein are essential for binding to ACE2. Cleavages at the S1-S2 boundary are a requirement for virus maturation, they occur in the trans-Golgi network of the viral-producer host cell, and are carried out by Ca2+-dependent proprotein convertase – furin (Johnson et al., 2020). Cleavage on S2’ site occurs during the viral entry, and it is associated with target-cell proteases. Depending on the type of viral entry pathway, two major proteases are involved in the cleavage: cathepsin L in endosomal entry pathway and transmembrane protease, serine 2 (TMPRSS2) in the cell surface entry pathway (Figure 2). Inhibition of furin or/and TMPRSS2 results in deactivation of the S protein (Shapira et al., 2022; Essalmani et al., 2022).
The engagement of spike protein with ACE2 for viral fusion and entry is a complex multistep process. First, the RBD changes its conformation to a slightly open state that allows binding to the receptor. This in return exposes S2’ site as the protein refolds and HR1 thrusts the cell membrane which inserts FP. The following dissociation of S1 causes the folding back of HR2 that leads to the juxtaposition of FP to the transmembrane region and the fusion of the membranes. The process continues with the formation of the fusion pore by the same HR2 leading to the facilitation of viral entry (Jackson et al., 2022).
ACE2 is the main cellular receptor for coronaviruses SARS-CoV-2, SARS-CoV, and HCoV-NL63 (Hofmann et al., 2005; Jackson et al., 2022; Beyerstedt et al., . 2021; Li et al., 2007). It has been found that early trypsin priming may enhance SARS-CoV-2 infection in cultured cells (Kim et al., 2022). Similarly, an increase in the infectivity of SARS-CoV is seen in the presence of proteases such as trypsin, elastase and thermolysin (Matsuyama et al., 2005). Various receptor molecules are also known to facilitate the penetration and infection of the SARS-CoV-2 virus (Alipoor and Mirsaeidi, 2022; Mollentze et al., 2022; Xia, 2023), such as neuropilin receptors (Daly et al., 2020; Cantuti-Castelvetri et al., 2020), C-lectin type receptors, dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin, Liver/lymph node-specific intercellular adhesion molecule-3-grabbing integrin, Macrophage Galactose-type Lectin (Thépaut et al., 2021; Lempp et al., 2021), glucose-regulated protein (Carlos et al., 2021; Shin et al., 2022), heparan sulfate proteoglycans (Zhang et al., 2023; Kearns et al., 2022; Bermejo-Jambrina et al., 2021), AXL Receptor Tyrosine Kinase (Wang et al., 2021). Another receptor molecule that also facilitates viral entry is CD147. Ragotte RJ et al. showed that CD147-mediated facilitation is not via binding to the RBD region of the virus (Ragotte et al., 2021).
The principle of the viral entry in MERS-CoV is similar in terms of the process to the SARS-CoV. However, a major distinct feature is a different receptor used by the virus for attachment and entry. Human dipeptidyl peptidase 4, type II transmembrane ectopeptidase serves as a receptor for MERS-CoV (Meyerholz et al., 2016). Depending on the tissue type and host cell, entry of MERS-CoV into cells can occur either through fusion or via endosomes. Experiments by Qian et al. carried out on VERO E6 and 293T cell lines show that if the MERS-CoV spike protein on pseudovirions is not digested by trypsin or TMPRSS2/4 proteases, then the viruses enter through endocytosis in a cathepsin L-dependent manner. However, if the MERS-CoV S protein is cleaved either during virus maturation by proteases or by trypsin in the extracellular fluid, the viruses penetrate the plasma membrane at neutral pH. This induces syncytia formation even in cells that express low or no levels of the MERS-CoV receptor (Qian et al., 2013).
HCoV-OC43 utilizes sialic acids, specifically N-acetyl-9-O-acetylneuraminic acid as an attachment receptor to bind to the host cell surface. This is similar to the bovine coronavirus, suggesting zoonotic origins for OC43. Unlike many other human coronaviruses which employ receptors like aminopeptidase N or angiotensin-converting enzyme 2, OC43 was found to use either HLA class I molecules or sialic acids as its fusion receptor for cellular entry.
Upon receptor binding, OC43 initiates a caveolin-mediated endocytic pathway for internalization. The virus particles go to caveolae, which are flask-shaped invaginations in the cell membrane containing caveolin-1. This caveolar route allows OC43 cellular entry in a manner distinct from other coronaviruses that primarily use clathrin-mediated endocytosis. While actin filaments are not directly required, unwinding of the actin cortex at the cell surface seems necessary to facilitate OC43 receptor binding and initial entry via caveolae-mediated endocytosis (Owczarek et al., 2018).
Similar to OC43, HCoV-HKU1 uses sialic acids for binding to the surface membrane of the host cell (Liu et al., 2021). However, Saunders et al. (2023) showed that TMPRSS2 acts as a receptor for HCoV-HKU1, facilitating viral entry in two ways. First, its enzymatic activity primes the virus for membrane fusion at the cell surface. Second, even when TMPRSS2 is inactive, it can still bind the virus, allowing entry through endosomes. This dual role suggests TMPRSS2 is crucial for HKU1 entry and a potential target for antiviral strategies (Saunders et al., 2023).
The most probable receptor for HCoV-229E is human aminopeptidase N (hAPN) which is identical to CD13, a glycoprotein of monocytes, granulocytes and their progenitors (Yeager et al., 1992).
2.2.2 Cellular effects of the S proteinThe numerous studies show that spike protein initiate ER stress induction and activation of unfolded protein response, which in turn leads to innate immune response, microRNA modulation, autophagy and cell death (Xue and Feng, 2021; Versteeg et al., 2007; Chan et al., 2006).
It was investigated whether the SARS-CoV-2 spike protein could prime the NLRP3 inflammasome in microglia cells, in addition to directly activating it. The cells were exposed to the spike protein, and induction of priming of inflammasomes through NF-κB signaling was observed. Priming would make the cells more responsive to subsequent inflammasome triggers. When ATP or nigericin were added following spike protein exposure, higher IL-1β release was observed compared to the control without prior spike protein exposure. This showed that spike protein indeed induces inflammasome activation in microglial cells through NF-κB (Albornoz et al., 2023).
SARS-CoV-2 S protein engagement with the ACE2 receptor reduces the expression of ACE2 on the cell over time. It also can induce caspase activation with following apoptosis in endothelial cells. Spike protein reduces the production of KLF2 (Krüppel-like Factor 2) and increases the expression of vWF (von Willebrand factor) in primary human arterial endothelial cells. This in turn leads to vascular inflammation and coagulation due to endothelial cell dysfunction (Panigrahi et al., 2021). In HEK293 cells presence of spike protein results in syncytia formation and cell sloughing. The protein also induced TNF-α, MCP-1, and ICAM1 mRNA expression as well as of heme oxygenase-1 (Singh et al., 2022). It was also observed that S protein’s RBD of SARS-CoV-2 upregulates secretion of the IL-6 and IL-8 through ATP/P2Y2 and ERK1/2 signaling pathways in human bronchial epithelia (Zhang et al., 2024). Recombinant subunits of spike protein of SARS-CoV-2 contrary to the previous studies induce CXCL10 chemokine expression that is attenuated via glycogen synthase kinase-3 inhibitor, not through the NF-kB, but rather IRF transcription factor that is TLR2-independent in human macrophage cells (THP-1) (Ghazanfari et al., 2024). Through the Proliferating Cell Nuclear Antigen (PCNA) expression it was identified that S protein of SARS-CoV-2 suppresses cell proliferation of SiHa cell line. The significant increase of expression of anti-proliferative p53 molecule is a suggested mechanism for cellular apoptosis along with pro-apoptotic TRAIL (TNF-related apoptosis-inducing ligand) that is also upregulated (Willson et al., 2024).
The findings of recent research by Monaco et al. (2024) suggest that the S1 domain of spike protein inhibits lactate dehydrogenase B which converts lactate to pyruvate through depletion of NAD+. Such inhibition shifts the metabolism from aerobic to anaerobic pathways. This shift is similar to the Warburg Effect, observed in viral infections and cancers, with cells relying more on glycolysis despite abundance of oxygen. Along with that, the upregulation of proteins contributing to Warburg Effect such as hexokinase-2 (HK2), hypoxia up-regulated protein 1(HYOU1), and TBC1 domain family member 4 (TBC1D4) in the HEK-293T transfected cell line with S1 domain of the S protein was observed in comparison to the control (Monaco et al., 2024).
Another novel finding regarding alternative binding of S protein to β1- and β2-ARs in cardiomyocytes suggests that the virus contributes to cardiac dysfunction which is observed in post-acute sequelae cardiovascular syndrome (PASC-CVS) of COVID-19. Activation of those receptors increases cAMP accumulation in the downstream signaling, and leads to cardiac sympathetic hyperactivity and thus weakens heart function (Deng et al., 2024).
Incubated RAW 264.7 macrophages with truncated spike protein of SARS-CoV induced secretion of IL-6 and TNF-α cytokines through NF-κB pathway (Wang et al., 2007).
A single point mutation (Y241H) in the spike protein of HCoV-OC43 was shown to modulate virus-induced neuropathogenesis in mice, resulting in death. Mice infected with the recombinant virus bearing this mutation (rOC/US241) developed a motor paralysis syndrome with demyelination in the spinal cord, while the reference virus caused only encephalitis. rOC/US241 replicated at a similar levels as the reference virus in the brain but persisted longer in the spinal cord. The Y241H mutation led to neuronal dysfunction shown by abnormal neurofilament phosphorylation. It also downregulated the glutamate transporter GLT-1 in astrocytes and strongly activated microglia/macrophages compared to the reference virus. Treatment with an AMPA receptor antagonist reduced motor dysfunction in rOC/US241 infected mice by attenuating neuronal and glial alterations as well as microglial activation (Brison et al., 2011). The effects of the spike protein on a host cell are summarized in the Table 2.
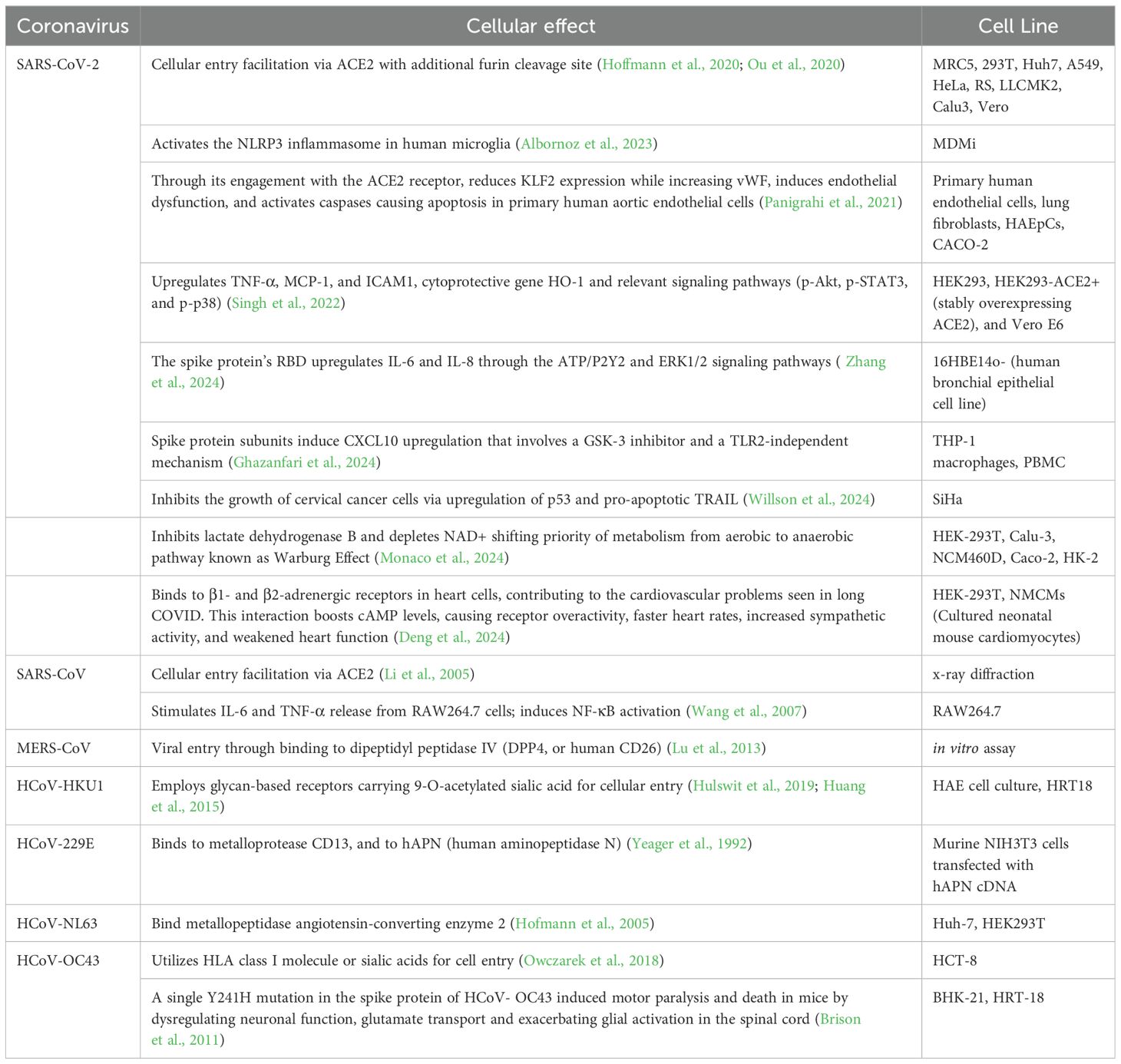
Table 2. Cellular effects of spike protein of human coronaviruses.
3 Structure and function of E protein3.1 StructureThe coronavirus envelope (E) protein is а short, integrаl membrаne protein consisting of 76 to 109 аmino аcids, with а size rаnging from 8.4 to 12 kDа (Schoeman and Fielding, 2019). Structurally, it comprises three distinct domаins, as shown in Figure 3. Amino (N)-terminal domain (NTD) is a short, hydrophilic region consisting of 7 to 12 amino acids. Transmembrane domain (TMD) is a large hydrophobic region of 25 amino acids, consisting of at least one predicted amphipathic α-helix. This domain enables the oligomerization of E proteins to form an ion-conductive pore across membranes. Carboxy (C)-terminal domain (CTD) is a hydrophilic region which makes up most of the protein. This domain contains β-coil-β motif, which functions as a Golgi-complex targeting signal (Nieto-Torres et al., 2015) (Figure 4A).
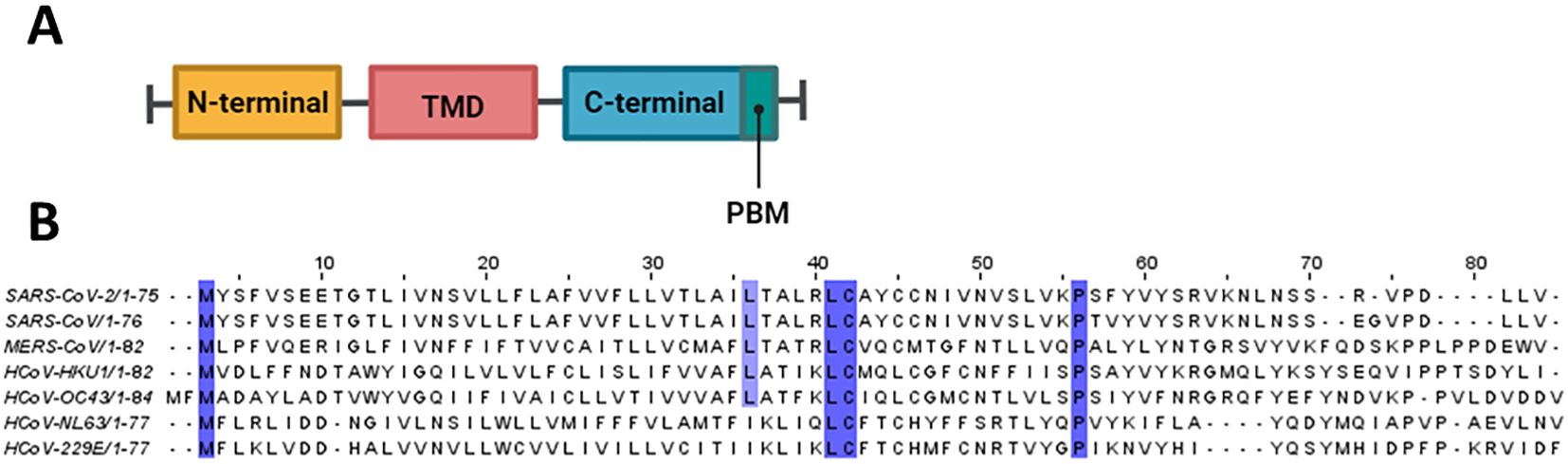
Figure 3. Structural features of E protein. (A) Schematic representation of coronavirus E protein structure. (B) Multiple sequence alignment was conducted using Clustal Omega and visualized in Jalview. Conserved residues are highlighted in purple.
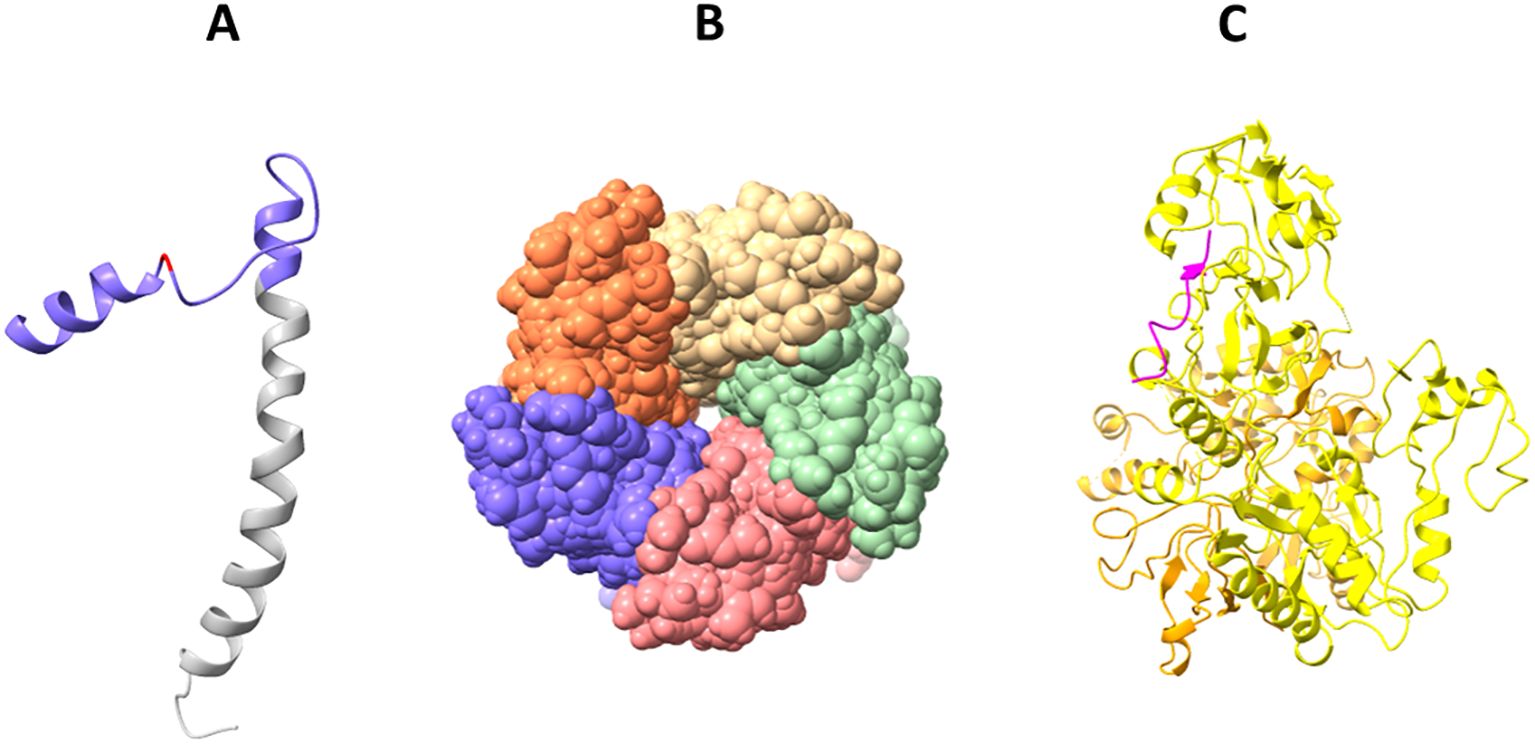
Figure 4. Solved 3D structures of coronavirus E protein. (A) SARS-CoV-2 E protein structure. Golgi complex-targeting signal located on the SARS-CoV CTD (colored in purple) is shown in pink, and the TMD is colored in gray (PDB:2MM4); (B) Pentameric structure of SARS-CoV-2 E protein transmembrane domain (PDB: 5X29); (C) Cryo-electron microscopy structure of SARS-CoV-2 E protein PBM (shown in magenta) interaction with host PALS1 (PDB: 7NTK).
The E protein in coronaviruses adopts an N-ecto/C-endo topology with one transmembrane domain. This topology implies that the N-terminus of the protein is located outside the virus (ectodomain), and the C-terminus is located inside the virus (endodomain). This single transmembrane domain is crucial for the integration of the E protein into the host cell membrane (Nieto-Torres et al., 2015; Schoeman and Fielding, 2019).
Solid-state NMR spectroscopy revealed that the transmembrane domain of the E protein of SARS-CoV-2 assembles into a homopentameric structure, forming a narrow pore within membranes (Mandala et al., 2020) (Figure 4B). However, the recently conducted NMR study found that it can exist as a dimer in lipid bilayers (Zhang et al., 2023). In addition, an ectodomain-containing E construct (ENTM, aa 1-41) from SARS-CoV-2 forms dimers instead of pentamers in lipid bilayers (Somberg et al., 2024). They found that oligomeric state and drug binding of the E protein is affected by the presence of ectodomain. This research clearly demonstrates that E protein may adopt different oligomeric states, and depending on that the binding of antivirals may be affected. It is therefore hypothesized that E proteins using different oligomeric states perform different functions, which are to be investigated in future studies.
The CTD of the SARS-CoV-2 E protein contains a PDZ-binding motif (PBM), which is crucial for establishing interactions with host cell proteins. This PBM in the CTD of the SARS-CoV-2 E protein interacts with host cell junction proteins such as PALS1 (Chai et al., 2021) (Figure 4C). This interaction induces relocation of PALS1 from the cell junction to the endoplasmic reticulum–Golgi intermediate compartment (ERGIC), where viral assembly and maturation take place (Chai et al., 2021). One study found that amino acid variations within the CTD of SARS-CoV-2 E protein, notably at residues Ser 55 -Phe 56, Arg 69, and the C-terminal end (DLLV: 72-75), may alter its binding affinity to PALS1 (Rahman et al., 2021).
Cellular studies showed that SARS-CoV-2 E protein PBM interacts with syntenin and ZO1 (Ávila-Flores et al., 2023). Host cell proteins associated with cellular junction and polarity such as TJP1, PARD3, MLLT4, LNX2 interact with the E protein’s PBM, leading to the sequestration of these PDZ domains to the Golgi compartment (Zhu et al., 2022). All these findings show that the coronavirus uses its E protein to disturb cellular communication and integrity, thereby enabling viral propagation.
Like in SARS-CoV-2, the TM domain of the SARS-CoV E protein forms a pentameric ion channel across membranes. It was shown that leucine and valine rich region within the SARS-CoV E protein TM domain is critical for the formation of the ion channel. Additionally, SARS-CoV E protein possesses a triple cysteine motif, which interacts with the spike protein of the virus (Aldaais et al., 2021).
The MERS-CoV E protein has a single α-helical transmembrane domain (Surya et al., 2015). This transmembrane domain can form pentameric ion channels in membranes. Similar to the E proteins of SARS-CoV and SARS-CoV-2, the MERS-CoV E protein also has a C-terminal PBM. However, unlike the E proteins from SARS-CoV and SARS-CoV-2, the PBM of MERS-CoV E protein does not interact with the host cell protein PALS1 (Javorsky et al., 2021).
There was a recent study focused on the determination of the structural properties of NL63 (Sučec et al., 2024). The study found that the TMD of the E protein (ETM) of NL63 adopts an α-helical conformation. Interestingly, they found that upon pH decrease or the presence of Ca2+ ions, the ETM of NL63 does not show much change in its water accessibility, whereas the water accessibility of the SARS-CoV-2’s ETM increases upon the same conditions. These functional differences can be attributed to the structural differences between the two viruses. As discussed in the paper, NL63 ETM possesses a 7DDN9 motif, which compared to the corresponding motif (7EET9) present in the SARS-CoV ETM, is less able to respond to the changes in pH and Ca2+ ions due to differences in sidechain charge and lengths. As shown in sequence alignment in Figure 3, 7EET9 motif is present in SARS-CoV and SARS-CoV-2, while missing in less pathogenic HCoVs (HKU1, OC43,NL63, 229E). The previous structural studies conducted on the SARS-CoV ETM identified three Phe residues positioned three residues apart from each other in its hydrophobic segment, which played a role in the channel’s gating function. In the case of the NL63 ETM, three Phe residues are positioned successively, and thus may be unable to participate in gating function. In Figure 3, it is shown that three Phe residues positioned three residues apart from each other are common to SARS-CoV and SARS-CoV-2, whereas less pathogenic HCoVs do not share this feature. The experimental conditions used with low pH and high Ca2+ concentrations highly resemble the conditions in the ERGIC compartment, in the membrane of which E protein is located. Thus, lower pathogenicity of coronaviruses can partly be linked to their reduced viroporin activity.
From the sequence alignment in Figure 3, we can see that M residue located in the NTD and L, C and P residues located in the CTD of the E protein are conserved across all HCoVs. Additionally, three Phe residues positioned three residues apart from each other are common to SARS-CoV and SARS-CoV-2 (F22, F25, F28) whereas less pathogenic HCoVs do not share this feature. This may partly explain the enhanced pathogenicity of SARS-CoV and SARS-CoV-2 in relation to other less virulent HCoVs.
3.2 FunctionWhen compared to other coronavirus structural proteins, the E protein is unique in the sense that only a small proportion of it forms virions, while most gets incorporated into the membrane of the ERGIC in infected cells (Venkatagopalan et al., 2015). Although research on the coronavirus E protein is quite scarce, based on the available data, it is certain that the E protein plays crucial roles in the virus’s lifecycle (Figure 5; Table 3).
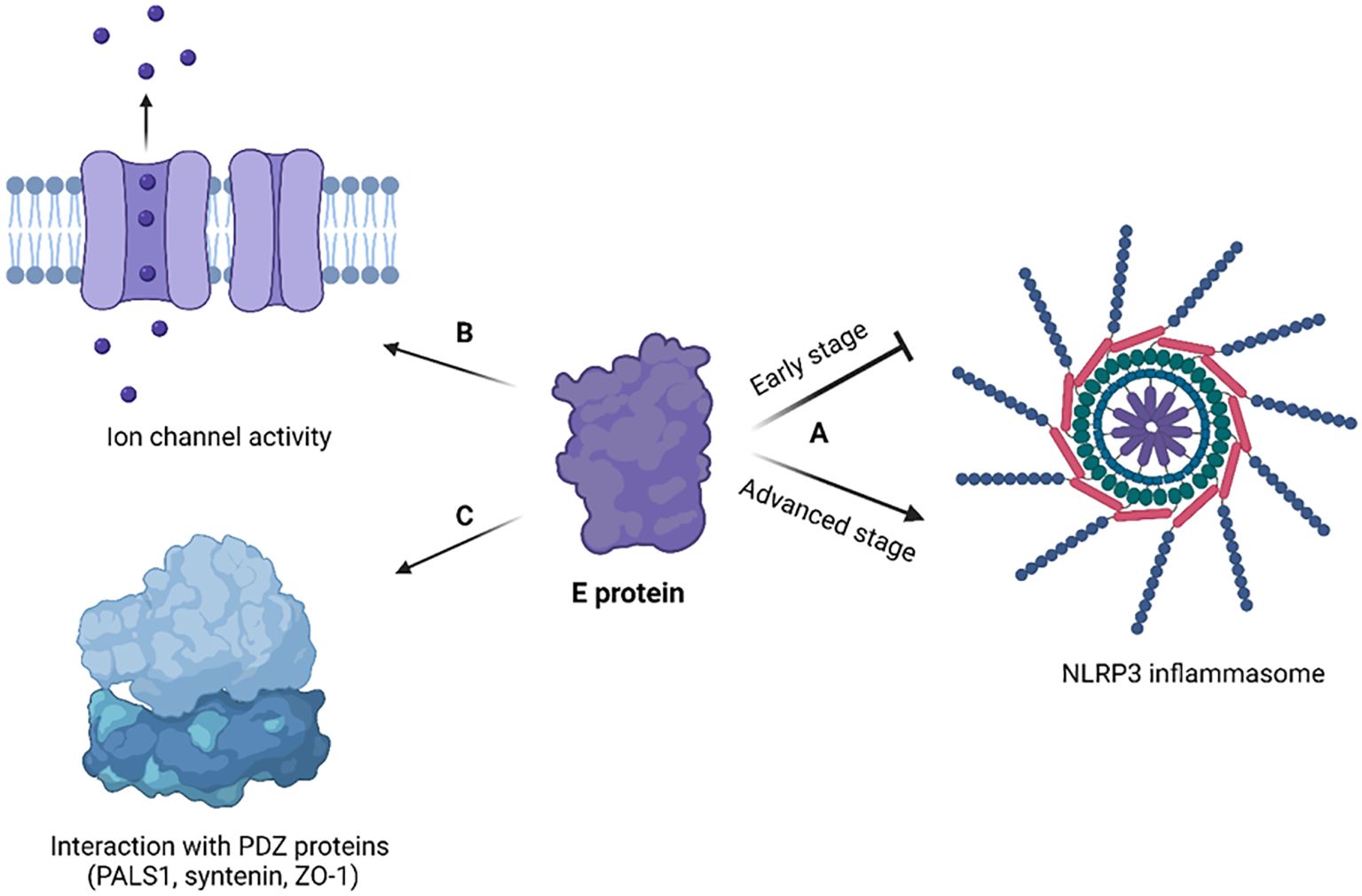
Figure 5. Cellular effects of coronavirus E protein. (A) In early stage of disease, E protein dampens the activation of NLRP3 inflammasome, while in advanced stage it activates NLRP3 inflammasome and exacerbates immune response; (B) E protein has ion channel activity and enables Ca2+ ions transport across ERGIC membrane; (C) E protein interacts with human PDZ proteins (PALS1, syntenin, ZO-1) through its PBM. The figure is created using BioRender.
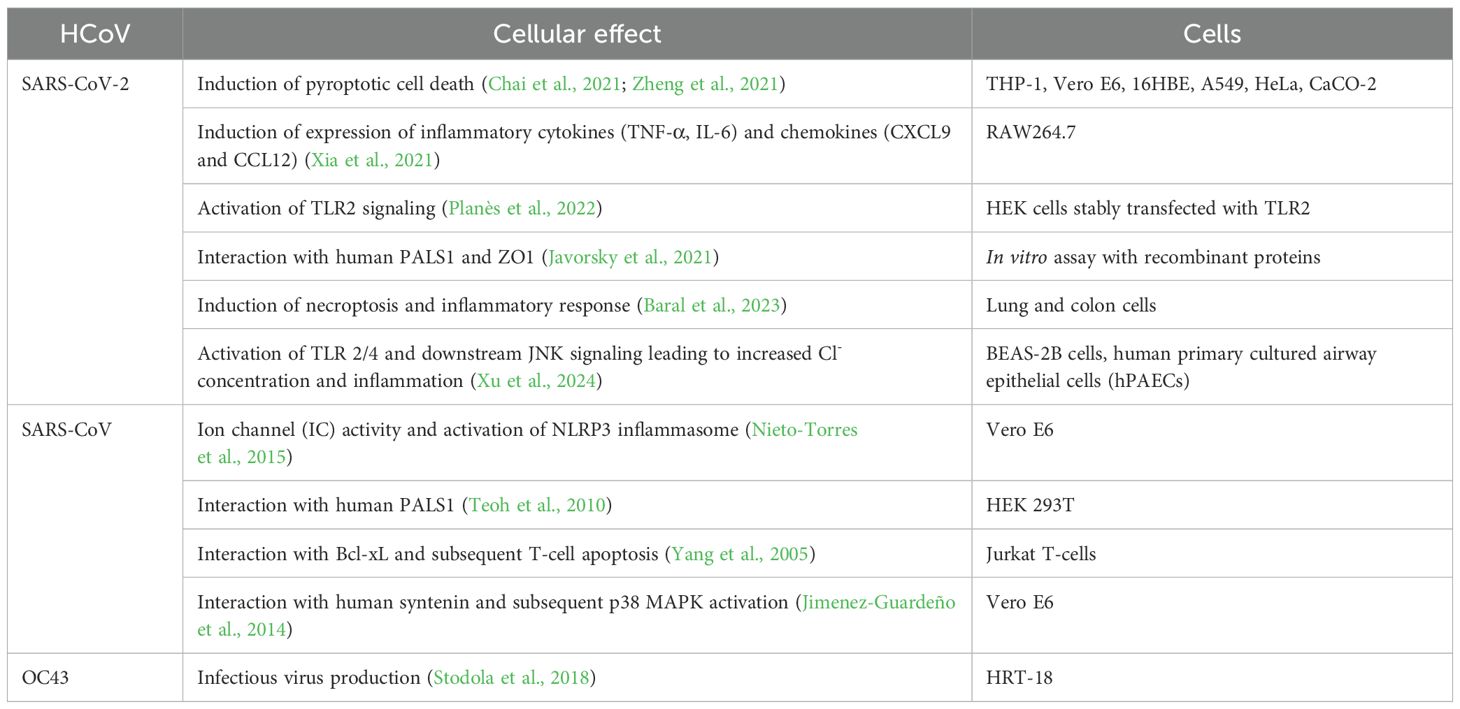
Table 3. Cellular effects of human coronavirus E protein.
3.2.1 Virus assembly and buddingThe viral E protein of SARS-CoV-2 plays a crucial role in retaining the S protein inside infected cells, specifically localizing it to the membranes of the ERGIC (Endoplasmic Reticulum-Golgi Intermediate Compartment) or the Golgi apparatus by slowing down the host cell’s secretory pathway (Boson et al., 2021). Furthermore, the E protein, in collaboration with M protein, facilitates the N-glycosylation of the S protein through a mechanism that operates independently of its intracellular retention (Boson et al., 2021). This coordinated action between the E, M, and S proteins is essential for the proper assembly of virus-like particles.
The E protein, in conjunction with the M protein, facilitates the budding of the virus within the ERGIC. In a study, atomistic molecular dynamics simulations were conducted to understand this process better (Collins et al., 2021). The simulations utilized refined structural models of the SARS-CoV-2 M protein dimer and E protein pentamer. The results showed that while multiple M protein dimers acted together to induce global membrane curvature through protein-lipid interactions, the E protein pentamers helped to keep the membrane planar. This cooperation between the E and M proteins is fundamental for the budding process to occur effectively.
Another study found that the monomeric E protein both generates and senses membrane curvature, preferring to localize with its C-terminus at the convex regions of the membrane (Kuzmin et al., 2022). This characteristic is also observed when the protein is in its pentameric form. The localization to curved regions is deemed favorable for the assembly of E protein oligomers, and the induction of curvature is suggested to facilitate the budding of viral particles.
In summary, additional research is required to ascertain if the SARS-CoV-2 E protein can directly cause membrane curvature. Nonetheless, the role of the E protein in budding is unequivocal, particularly for its CTD.
3.2.2 Host cell effectsXia et al. showed that SARS-CoV-2 E protein forms a pH-sensitive cation channel causing cell death resembling pyroptosis (Xia et al., 2021). In addition, the E protein was shown to provoke robust immune responses, notably upregulating cytokines (TNF-α, IL-6) and chemokines (CXCL9, CCL12) both in vitro and in vivo, mirroring the cytokine storm observed in COVID-19 patients (Xia et al., 2021).
SARS-CoV-2 E protein transfection triggered necrotic cell death and inflammatory response in both lung and colon cells (Baral et al., 2023). The cellular effects of E protein have been mediated by the activation of the receptor interacting protein kinase 1 (RIPK1), which is a necroptotic marker. Subsequently, RIPK1 promotes the phosphorylation of NF-κB, a key transcription factor involved in inflammation. Recently, there was an intriguing finding that SARS-CoV-2 E protein switches the innate immune system to a tolerant state upon secondary infections (Geanes et al., 2024). Though initially E protein activates the innate immune system via its interaction with TLR-2, its long-term effect is to make monocytes and macrophages unresponsive to pathogens, contributing to immune dysregulation. This could potentially explain why patients with severe forms of COVID-19 are susceptible to secondary infections. SARS-CoV-2 E protein has been shown to activate TLR-2/4 and subsequently JNK signaling, which leads to a high intracellular Cl- concentration through increased expression of phosphodiesterase 4D (PDE4D) (Xu et al., 2024). The increased level of Cl- ions further drive inflammation by enhancing the phosphorylation of serum/glucocorticoid regulated kinase 1 (SGK1) (Xu et al., 2024). Interestingly, blocking SGK1 or PDE4D helps to mitigate the inflammatory response triggered by E protein. This highlights novel therapeutic targets to treat COVID-19 related inflammation.
Previous research on SARS-CoV showed that strains lacking the E protein couldn’t activate the NF-κB pathway, reducing inflammatory cytokine production (DeDiego et al., 2014). Recent studies on SARS-CoV-2 confirmed this finding, emphasizing the significant role of viral E in eliciting robust immune responses both in vitro and in vivo (Xia et al., 2021).
The SARS-CoV-2 E protein can induce the release of inflammatory cytokines like TNF-α and IFN-γ, and activate the NLRP3 inflammasome (Zheng et al., 2021). This activation is linked to the ion channel property of the E protein which facilitates ion transport, providing an activation signal for the NLRP3 inflammasome assembly. Moreover, the interaction between the SARS-CoV-2 E and Toll-like receptor 2 (TLR2) was identified, which further underscores the role of E protein in innate immune responses. The modulation of the NLRP3 inflammasome by E protein varies across different infection stages (Yalcinkaya et al., 2021). Initially, it may suppress the host NLRP3 inflammasome response to viral RNA but might enhance the NLRP3 inflammasome response in later infection stages.
The E protein disrupts cell polarity by interacting with certain connexins. The interaction primarily occurs at a site known as the PDZ-Binding Motif (PBM) in the E protein, which comprises the last four carboxy-terminal amino acids (DLLV) (Chai et al., 2021). PDZ domains are common protein interaction modules that recognize short amino acid sequences at the C-terminus of target proteins. These domains are found in a variety of connexins, and they can recognize and interact with several human cell junction proteins including PALS1, ZO-1, and syntenin. PALS1 is a crucial protein associated with tight junctions and plays a vital role in maintaining epithelial polarity. Interaction of SARS-CoV E protein with PALS1 has been associated with lung epithelial cell destruction in SARS patients (Teoh et al., 2010). Comparatively, the E protein of SARS-CoV-2 showed an increased affinity for PALS1’s PDZ domain which could be a contributing factor to SARS-CoV-2’s increased virulence (Toto et al., 2020).
A study by Chai et al. used cryo-electron microscopy to visualize the complex structure formed by PALS1 and SARS-CoV-2 E protein, revealing how the DLLV motif of E protein recognizes a hydrophobic pocket formed by PDZ and SH3 domains of PALS1 (Chai et al., 2021). This interaction disrupts the apical cell polarity complex, which could lead to loosened and leaky lung epithelial junctions, promoting local viral spread and immune cell infiltration into lung alveolar spaces.
4 Structure and function of M protein4.1 StructureThe M protein, being the predominant structural protein of the virus, plays a critical role in driving the assembly of the virus and initiating the budding process from the membrane (Z. Zhang et al., 2022). Structurally it consists of a short exterior N-terminal domain (NTD), three transmembrane domains and a long C-terminal domain, located inside the virion, as shown in Figure 6.
Comments (0)