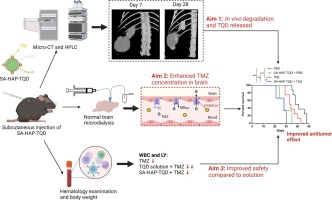
Respiratory pathologies, including chronic obstructive pulmonary disease (COPD), lower respiratory infections and lung cancer, are among the top five leading causes of death. Current state-of-the-art therapies often provide insufficient disease control. In addition, the global prevalence of lung disease is expected to further increase, leading to a substantial economic and public health-related societal burden [1], [2]. Hence, there is an urgent need for the development of novel, disease-modifying therapies.
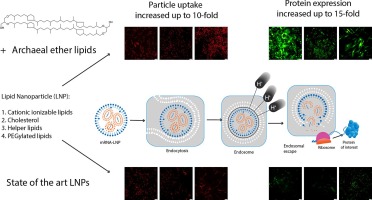
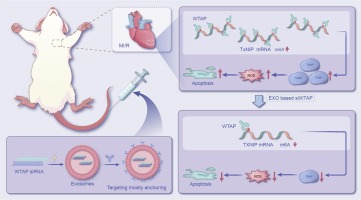
Over the last two decades, there has been a growing interest in RNA interference (RNAi) therapy. Activation of the cytosolic RNAi pathway by small interfering RNA (siRNA) allows sequence-specific silencing of target genes on the post-transcriptional level. Importantly, as in theory all human genes are susceptible to RNAi, siRNA drugs have the potential to alleviate the large unmet medical need for complex difficult-to-treat pathologies [3], [4], [5], [6]. Currently, the liver is the target organ of choice for systemic siRNA therapies, as exemplified by the initial clinical approval of an siRNA-loaded lipid nanoparticle (LNP) formulation (patisiran) in 2018 and more recently several GalNac-siRNA conjugates (givosiran, lumasiran, inclisiran and vutrisiran) [6], [7]. On the other hand, targeted delivery of siRNA drugs to extrahepatic tissues could considerably broaden therapeutic applicability. Local pulmonary administration of siRNA is an attractive strategy to reduce the expression of disease-related genes in distinct lung cells [8], [9]. To overcome the various extra- and intracellular barriers towards its cytosolic mRNA target, siRNA is typically formulated into synthetic lipid- or polymer-based nanoparticles (NPs), which are adequately internalized by cells via endocytosis [10], [11]. However, the majority of currently designed NPs fails to efficiently induce escape of the encapsulated siRNA from the endosomal lumen into the cytosol. Consequently, internalized NPs are rapidly trafficked towards the lysosomes, where both the particle and its therapeutic cargo are prone to degradation, or expelled from the cell via endocytic recycling pathways. As a result, even state-of-the-art NPs allow only a fraction of the internalized siRNA dose to be released into the cytosol and eventually exert its biological effect [12], [13], [14], [15], [16].
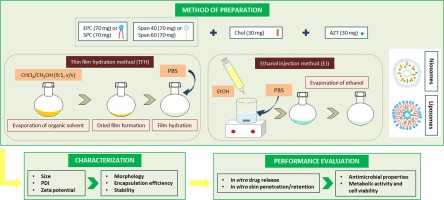
We previously developed hybrid NPs that consist of a cationic siRNA-loaded polymeric matrix core, which is electrostatically coated with an anionic proteolipid shell of pulmonary surfactant (PS) [17], [18], [19]. The core consists of a nanosized, cationic dextran-based hydrogel (nanogel; NG) with a proven high loading capacity for siRNA, efficient endocytosis and subsequent siRNA delivery in pulmonary cell types [20]. Coating the siRNA-loaded NGs (siNGs) with Curosurf® (poractant α), a clinically-approved PS, improved both siNG stability as well as intracytosolic siRNA delivery, for which the latter was shown to be mediated by the membrane-active properties of surfactant protein B (SP-B) [17], [18], [21], [22], [23], [24].
As an alternative endosomal escape strategy, we reported previously that cationic amphiphilic drugs (CADs) can be repurposed as low molecular weight adjuvants for siRNA delivery [25]. Their physicochemical properties (clogP > 3; pKa > 6) allow these compounds to accumulate in the acidified (endo)lysosomes via pH-dependent ion trapping. Here, many CADs can act as functional inhibitors of the membrane-associated acid sphingomyelinase (FIASMA) [26], [27]. As such, their lysosomotropic behavior has been linked to the transient induction of a lysosomal storage disease-like phenotype involving (phospho)lipidosis, lysosomal swelling and lysosomal membrane permeabilization (LMP) [28], [29], [30], [31]. We have demonstrated that the latter phenotype can be exploited to facilitate cytosolic siRNA delivery of different siRNA nanocarriers following application to transfected cells [15], [25], [32], [33].
β2-adrenergic receptor agonists (β2-agonists) are of particular interest to be repurposed as delivery adjuvants for pulmonary siRNA therapy. Because of their bronchodilating effect, these drugs are already in clinical use for the treatment of COPD and asthma (e.g., in combination with inhaled corticosteroids) via inhalation therapy. In addition, as β2-agonists have cationic amphiphilic properties they can potentially be considered to promote cytosolic siRNA delivery in lung-related target cells. It was demonstrated earlier that the long-acting β2-agonist (LABA) salmeterol promotes siRNA delivery by NGs in a lung epithelial cell line [25]. However, at this point it remains unclear if this adjuvant effect can be extrapolated to other β2-agonists, nanocarriers and lung-related cell types. Of particular interest is to evaluate the compatibility of β2-agonists with the previously established PS-coated NG platform.
To address this question, in this work both short acting and (ultra-)long acting β2-agonists are evaluated to boost siRNA delivery by both uncoated as well as PS-coated siNGs in vitro in an epithelial non-small cell lung cancer (NSCLC) cell model. Moreover, in vivo biodistribution data following intranasal administration in mice clearly indicated that (PS-coated) siNGs tend to accumulate in the alveolar macrophage compartment. As such, the β2-agonists’ adjuvant effect was likewise assessed on a macrophage cell model. A clear correlation was observed between the physicochemical properties of the β2-agonists and their propensity to improve siRNA delivery, with only the (ultra-)long acting molecules being functional as siRNA delivery enhancers. Improved siRNA-induced gene knockdown by LABAs was observed in both epithelial and macrophage cell models and for both uncoated and PS-coated siNGs, albeit with clear differences in overall potency and toxicity.



Comments (0)