In recent years, formulation-induced supersaturation in the gastrointestinal region has gained importance as a promising method to enhance the oral absorption of poorly soluble drugs. Supersaturating drug delivery systems (SDDS) create a high-energy state of supersaturation, sustaining the drug in this state for a physiologically relevant duration [1]. In the supersaturated state, the drug concentration in the solution exceeds its equilibrium solubility, leading to increased free drug concentration, which serves as a driver for passive intestinal absorption [2]. Various formulation approaches, including the co-solvent method, lipidic solution, nanocrystal, co-crystal, and amorphous solid dispersion can induce supersaturation [3]. However, the therapeutic potential of SDDS in improving drug bioavailability largely depends on the stability of the generated supersaturated state.
Phospholipid-based solid dispersions offer an effective means of dispersion of drug molecules in the amorphous form within the matrix of phospholipid. Monoacyl and diacyl phospholipids are primarily employed in the preparation of phospholipid dispersions using methods such as solvent evaporation [4], spray drying [5], and freeze-drying [6]. The amphiphilic nature of these phospholipids contributes to augmentation in drug solubilization in the gastrointestinal (GI) tract through the encapsulation of drug molecules within micelle structures. In the literature, significant disparities have been noted in terms of both solubility and permeability modulation when using phospholipid-based formulations. Particularly, the influence on permeation remains uncertain, as demonstrated by a meta-analysis of solidified phospholipid dispersion studies [7]. Recently, Fong and colleagues demonstrated that phospholipid dispersion stabilizes the amorphous state of celecoxib within the matrix and generates true supersaturation on the dissolution [8]. In accordance with Fick's First Law, the increased concentration (molecularly dissolved) resulting from generated supersaturation at the absorption site augments the drug flux across the intestinal barrier. Similarly, Jacobsen et al. demonstrated that the complexities of biopharmaceutical performance depend on a complex interplay between supersaturation and recrystallization tendency when there is a low phospholipid content present in the dispersion [9].
To harness the full potential of this supersaturated state, the increased concentrations have to be maintained for sufficient periods to allow for significant absorption. Hence, a polymer is introduced as a ternary component within the phospholipid dispersion in this study [10]. The polymer would not only aid in maintaining supersaturation but also ensure its physical stability during storage. However, the choice of an appropriate polymer in the supersaturable formulation is critical, as it determines the supersaturation holding capacity, dissolution behavior, and interaction with the drug and phospholipid, all of which affect product performance [11], [12]. In this study, we selected HPMCAS as the polymeric carrier due to its superior ability to initiate and maintain drug supersaturation in dispersion systems for drugs with diverse structures and physical properties [13], [14]. In addition to preventing drug recrystallization in both solution and solid states, the amphiphilic nature of HPMCAS allows it to associate with drugs in hydrophobic regions. Simultaneously, it allows for the creation of drug-rich sub-micron particles, also known as nanodroplets along with the hydrated nanosized colloidal structures in aqueous environments [13], [15]. The formation of drug-rich submicron particles in the presence of solubilizing excipients, such as polymers or surfactants, is believed to influence the mass transport of the drug to the region of epithelial cells due to changes in the thermodynamic drug activity [16], [17], [18], [19].
Aprepitant is a selective antagonist of human substance P/neurokinin 1 (NK 1) receptors, used to alleviate chemotherapy-induced emesis, nausea, and vomiting, along with addressing postoperative nausea and vomiting [20]. APT was selected as the model drug for this study due to its distinct characteristics. The presence of strong intermolecular interactions within its crystal structure results in limited aqueous solubility (typically ranging from 3 to 7 μg/mL) and dissolution. Thereby, solubility, dissolution, and permeability are the rate-limiting steps for the gastrointestinal absorption of APT [21]. Additionally, APT is subject to positive food effects, first-pass metabolism, and nonlinear pharmacokinetics with increased therapeutic doses [22]. To address the aforementioned hurdles, implementing a supersaturating formulation approach could serve as a potential strategy to enhance the oral absorption of APT.
Hence, the formulation strategy of using phospholipid-based solid dispersion would be highly intriguing, as it has the potential to generate and maintain supersaturation in the GI lumen. However, the knowledge of how supersaturation influences drug absorption is lacking, with limited reports in the literature on drug supersaturation-mediated absorption [10]. Therefore, this study aims to address this gap by demonstrating, for the first time, the supersaturation-mediated absorption of a brick dust molecule using a phospholipid/polymer-based dispersion approach. Additionally, this study seeks to provide a comprehensive understanding of the supersaturable formulation's attributes, which encompasses its solid-state properties and the underlying supersaturation mechanism. In the following sections, we will present findings on the impact of supersaturation on drug absorption, shedding light on the complexities of biopharmaceutical performance.

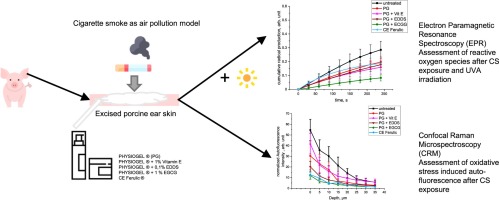
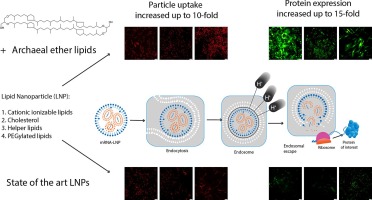

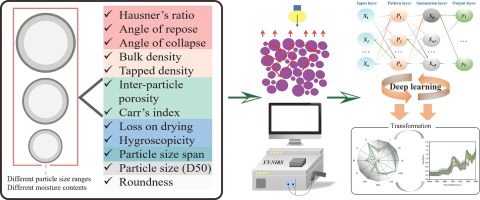
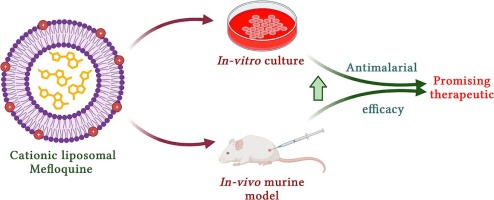

Comments (0)